

SWISS-MODEL: homology modelling of protein structures and complexes
- PMID: 29788355
- PMCID: PMC6030848
- DOI: 10.1093/nar/gky427
SWISS-MODEL: homology modelling of protein structures and complexes
Abstract
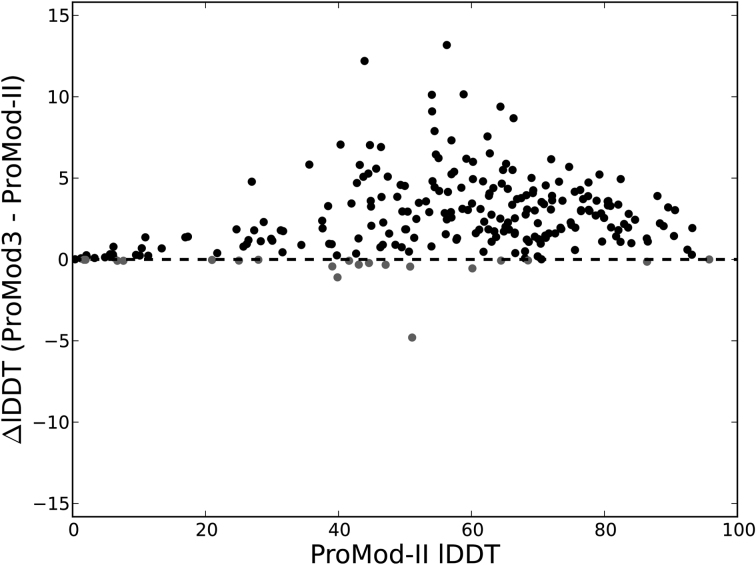
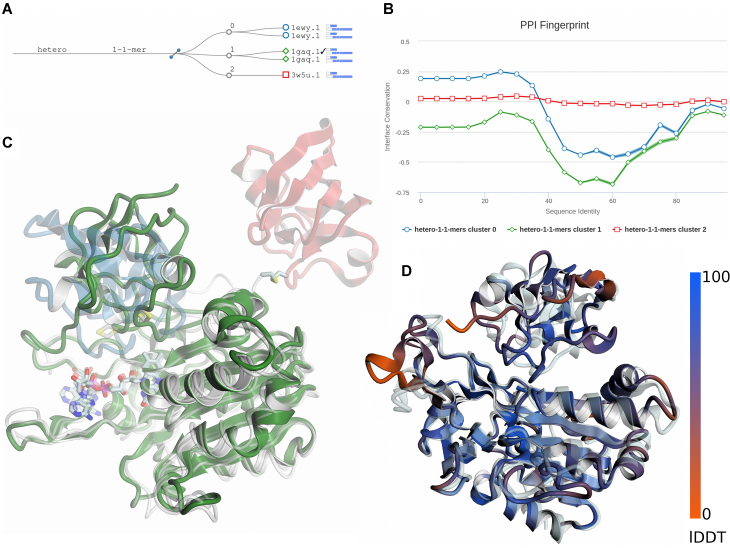
Homology modelling has matured into an important technique in structural biology, significantly contributing to narrowing the gap between known protein sequences and experimentally determined structures. Fully automated workflows and servers simplify and streamline the homology modelling process, also allowing users without a specific computational expertise to generate reliable protein models and have easy access to modelling results, their visualization and interpretation. Here, we present an update to the SWISS-MODEL server, which pioneered the field of automated modelling 25 years ago and been continuously further developed. Recently, its functionality has been extended to the modelling of homo- and heteromeric complexes. Starting from the amino acid sequences of the interacting proteins, both the stoichiometry and the overall structure of the complex are inferred by homology modelling. Other major improvements include the implementation of a new modelling engine, ProMod3 and the introduction a new local model quality estimation method, QMEANDisCo. SWISS-MODEL is freely available at https://swissmodel.expasy.org.
Figures
References
-
- Fuller J.C., Burgoyne N.J., Jackson R.M.. Predicting druggable binding sites at the protein-protein interface. Drug Discov. Today. 2009; 14:155–161. - PubMed
-
- Dutta S., Berman H.M.. Large macromolecular complexes in the Protein Data Bank: a status report. Structure. 2005; 13:381–388. - PubMed
-
- Marsh J.A., Teichmann S.A.. Structure, dynamics, assembly, and evolution of protein complexes. Annu. Rev. Biochem. 2015; 84:551–575. - PubMed
-
- Tramontano A. The computational prediction of protein assemblies. Curr. Opin. Struct. Biol. 2017; 46:170–175. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
