

Identification of new risk factors for rolandic epilepsy: CNV at Xp22.31 and alterations at cholinergic synapses
- PMID: 29789371
- PMCID: PMC6119347
- DOI: 10.1136/jmedgenet-2018-105319
Identification of new risk factors for rolandic epilepsy: CNV at Xp22.31 and alterations at cholinergic synapses
Abstract
Background: Rolandic epilepsy (RE) is the most common genetic childhood epilepsy, consisting of focal, nocturnal seizures and frequent neurodevelopmental impairments in speech, language, literacy and attention. A complex genetic aetiology is presumed in most, with monogenic mutations in GRIN2A accounting for >5% of cases.
Objective: To identify rare, causal CNV in patients with RE.
Methods: We used high-density SNP arrays to analyse the presence of rare CNVs in 186 patients with RE from the UK, the USA, Sardinia, Argentina and Kerala, India.
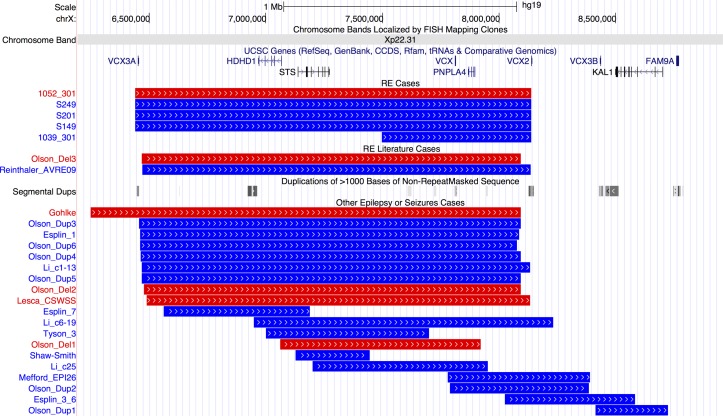
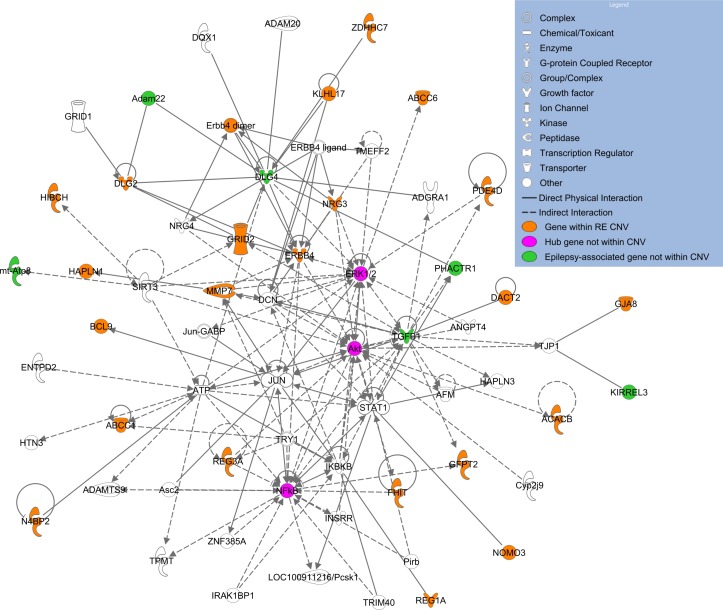
Results: We identified 84 patients with one or more rare CNVs, and, within this group, 14 (7.5%) with recurrent risk factor CNVs and 15 (8.0%) with likely pathogenic CNVs. Nine patients carried recurrent hotspot CNVs including at 16p13.11 and 1p36, with the most striking finding that four individuals (three from Sardinia) carried a duplication, and one a deletion, at Xp22.31. Five patients with RE carried a rare CNV that disrupted genes associated with other epilepsies (KCTD7, ARHGEF15, CACNA2D1, GRIN2A and ARHGEF4), and 17 cases carried CNVs that disrupted genes associated with other neurological conditions or that are involved in neuronal signalling/development. Network analysis of disrupted genes with high brain expression identified significant enrichment in pathways of the cholinergic synapse, guanine-exchange factor activation and the mammalian target of rapamycin.
Conclusion: Our results provide a CNV profile of an ethnically diverse cohort of patients with RE, uncovering new areas of research focus, and emphasise the importance of studying non-western European populations in oligogenic disorders to uncover a full picture of risk variation.
Keywords: copy-number; developmental; epilepsy and seizures; genome-wide.
© Article author(s) (or their employer(s) unless otherwise stated in the text of the article) 2018. All rights reserved. No commercial use is permitted unless otherwise expressly granted.
Conflict of interest statement
Competing interests: LA is a contractor for Eli Lilly and Company. SVT has received research grants from the Scientific bodies under the Government of India. DM consults for Cyberonics.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous