

Analysis of the genomic landscape of multiple myeloma highlights novel prognostic markers and disease subgroups
- PMID: 29789651
- PMCID: PMC6092251
- DOI: 10.1038/s41375-018-0037-9
Analysis of the genomic landscape of multiple myeloma highlights novel prognostic markers and disease subgroups
Abstract
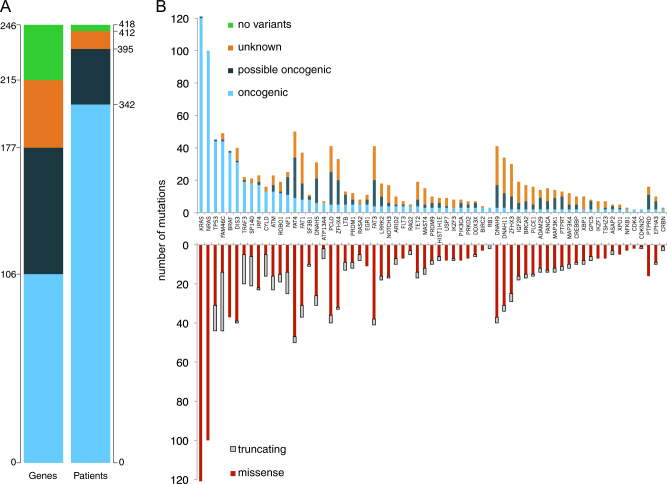
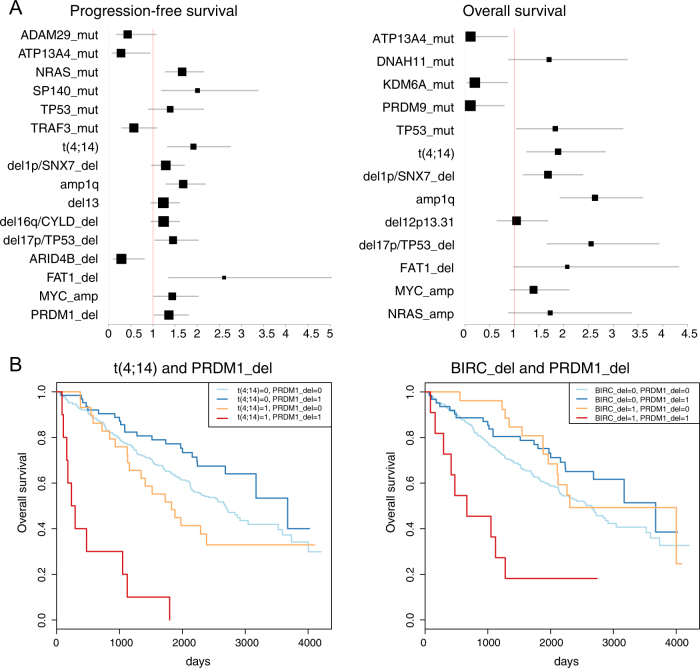
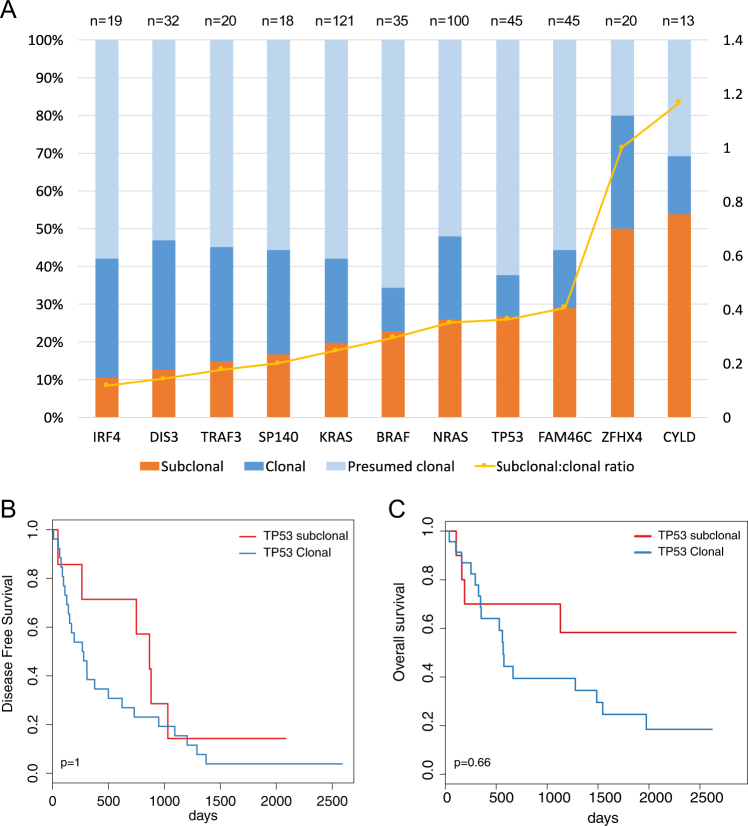
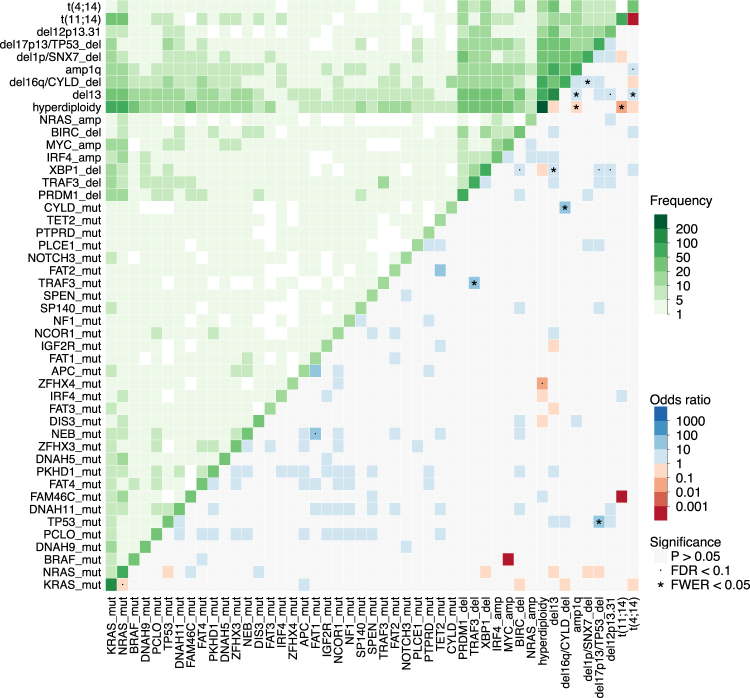
In multiple myeloma, next-generation sequencing (NGS) has expanded our knowledge of genomic lesions, and highlighted a dynamic and heterogeneous composition of the tumor. Here we used NGS to characterize the genomic landscape of 418 multiple myeloma cases at diagnosis and correlate this with prognosis and classification. Translocations and copy number abnormalities (CNAs) had a preponderant contribution over gene mutations in defining the genotype and prognosis of each case. Known and novel independent prognostic markers were identified in our cohort of proteasome inhibitor and immunomodulatory drug-treated patients with long follow-up, including events with context-specific prognostic value, such as deletions of the PRDM1 gene. Taking advantage of the comprehensive genomic annotation of each case, we used innovative statistical approaches to identify potential novel myeloma subgroups. We observed clusters of patients stratified based on the overall number of mutations and number/type of CNAs, with distinct effects on survival, suggesting that extended genotype of multiple myeloma at diagnosis may lead to improved disease classification and prognostication.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures


References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
