

Traceability, reproducibility and wiki-exploration for "à-la-carte" reconstructions of genome-scale metabolic models
- PMID: 29791443
- PMCID: PMC5988327
- DOI: 10.1371/journal.pcbi.1006146
Traceability, reproducibility and wiki-exploration for "à-la-carte" reconstructions of genome-scale metabolic models
Abstract
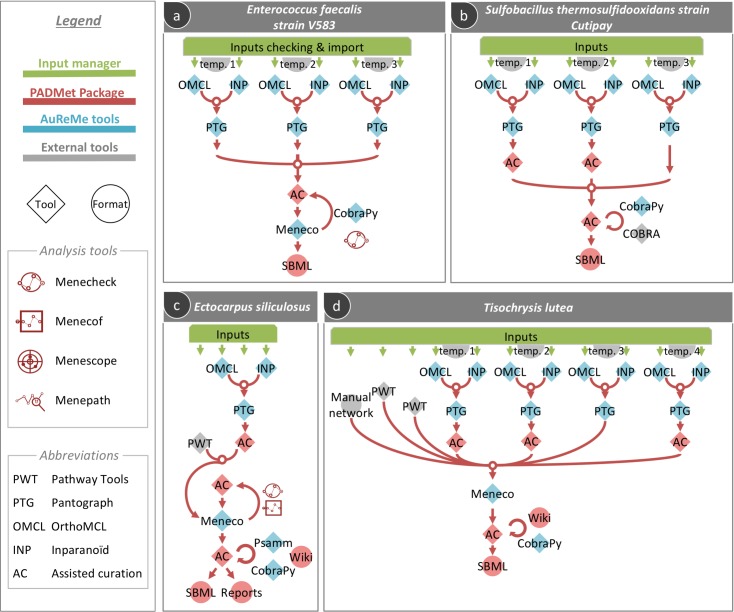
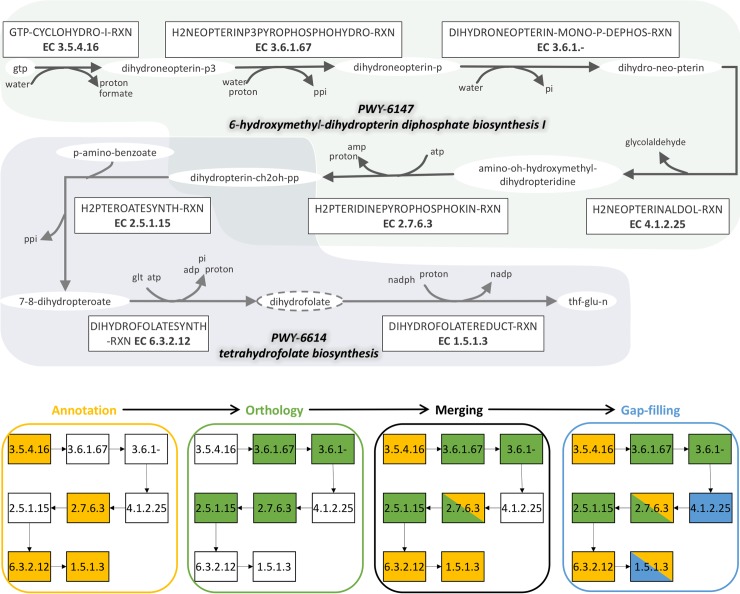
Genome-scale metabolic models have become the tool of choice for the global analysis of microorganism metabolism, and their reconstruction has attained high standards of quality and reliability. Improvements in this area have been accompanied by the development of some major platforms and databases, and an explosion of individual bioinformatics methods. Consequently, many recent models result from "à la carte" pipelines, combining the use of platforms, individual tools and biological expertise to enhance the quality of the reconstruction. Although very useful, introducing heterogeneous tools, that hardly interact with each other, causes loss of traceability and reproducibility in the reconstruction process. This represents a real obstacle, especially when considering less studied species whose metabolic reconstruction can greatly benefit from the comparison to good quality models of related organisms. This work proposes an adaptable workspace, AuReMe, for sustainable reconstructions or improvements of genome-scale metabolic models involving personalized pipelines. At each step, relevant information related to the modifications brought to the model by a method is stored. This ensures that the process is reproducible and documented regardless of the combination of tools used. Additionally, the workspace establishes a way to browse metabolic models and their metadata through the automatic generation of ad-hoc local wikis dedicated to monitoring and facilitating the process of reconstruction. AuReMe supports exploration and semantic query based on RDF databases. We illustrate how this workspace allowed handling, in an integrated way, the metabolic reconstructions of non-model organisms such as an extremophile bacterium or eukaryote algae. Among relevant applications, the latter reconstruction led to putative evolutionary insights of a metabolic pathway.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures



References
-
- Bordbar A, Monk JM, King ZA, Palsson BO. Constraint-based models predict metabolic and associated cellular functions. Nat Rev Genet. 2014;15: 107–20. doi: 10.1038/nrg3643 - DOI - PubMed
-
- Orth J, Thiele I, Bernhard. What is flux balance analysis? Nat Biotechnol. 2010;28: 245–248. doi: 10.1038/nbt.1614 - DOI - PMC - PubMed
-
- Yim H, Haselbeck R, Niu W, Baxley CP, Burgard A, Boldt J, et al. Metabolic engineering of Escherichia coli for direct production of 1,4-butanediol. Nat Chem Biol. 2011;7: 445–452. doi: 10.1038/nchembio.580 - DOI - PubMed
-
- Kim HU, Kim SY, Jeong H, Kim TY, Kim JJ, Choy HE, et al. Integrative genome-scale metabolic analysis of Vibrio vulnificus for drug targeting and discovery. Mol Syst Biol. John Wiley & Sons, Ltd; 2011;7: 460 doi: 10.1038/msb.2010.115 - DOI - PMC - PubMed
-
- Zelezniak A, Andrejev S, Ponomarova O, Mende DR, Bork P, Patil KR. Metabolic dependencies drive species co-occurrence in diverse microbial communities. Proc Natl Acad Sci U S A. 2015;112: 6449–6454. doi: 10.1073/pnas.1421834112 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
