

Acute lung injury: how to stabilize a broken lung
- PMID: 29793554
- PMCID: PMC5968707
- DOI: 10.1186/s13054-018-2051-8
Acute lung injury: how to stabilize a broken lung
Abstract
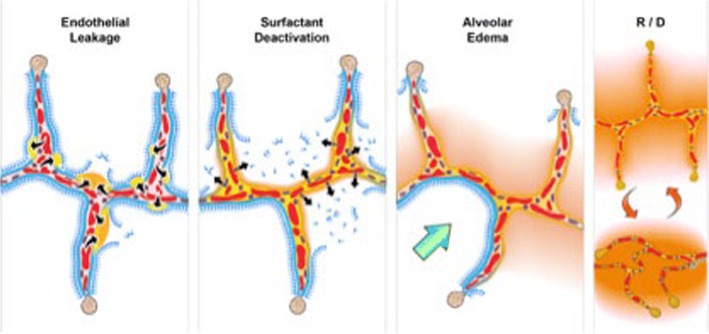
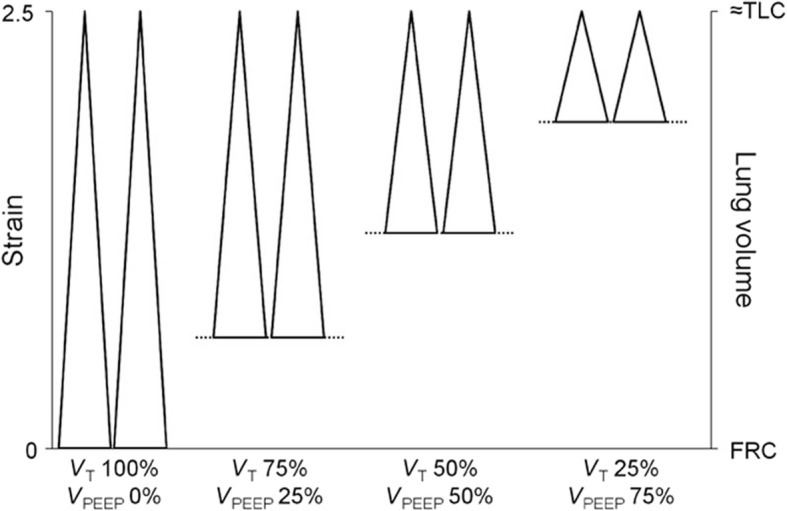
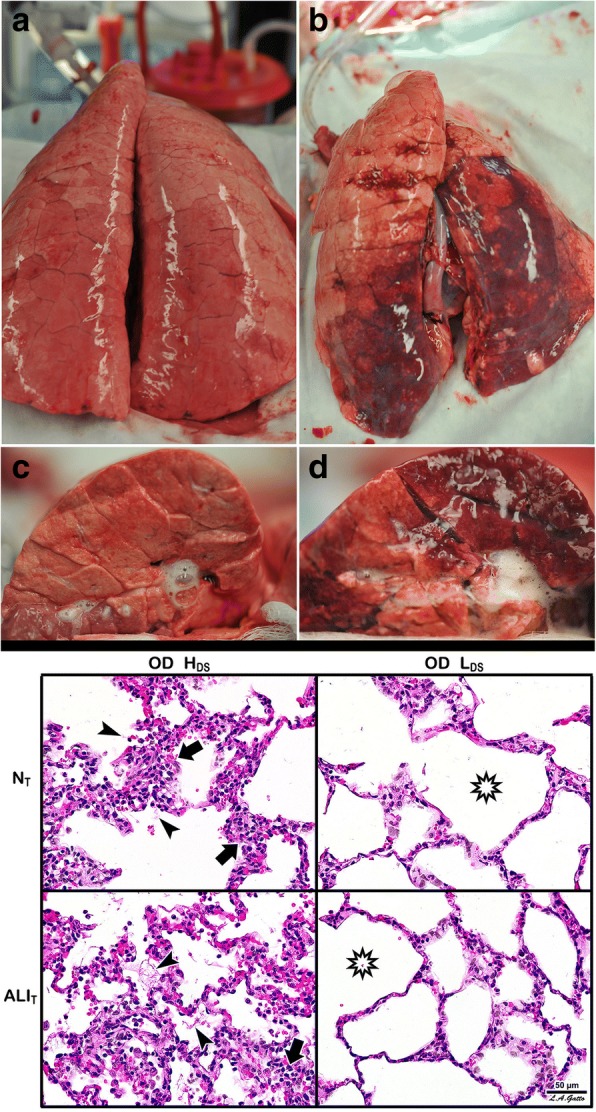
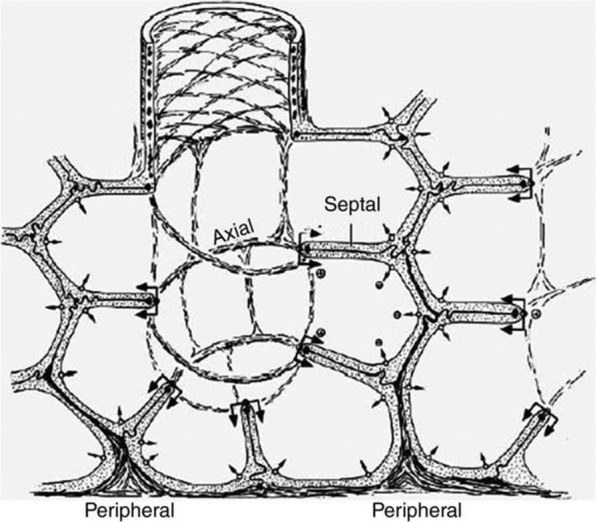
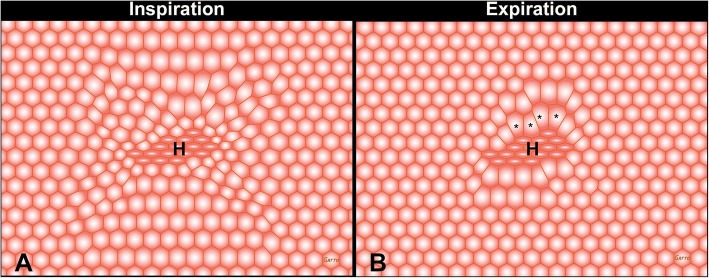
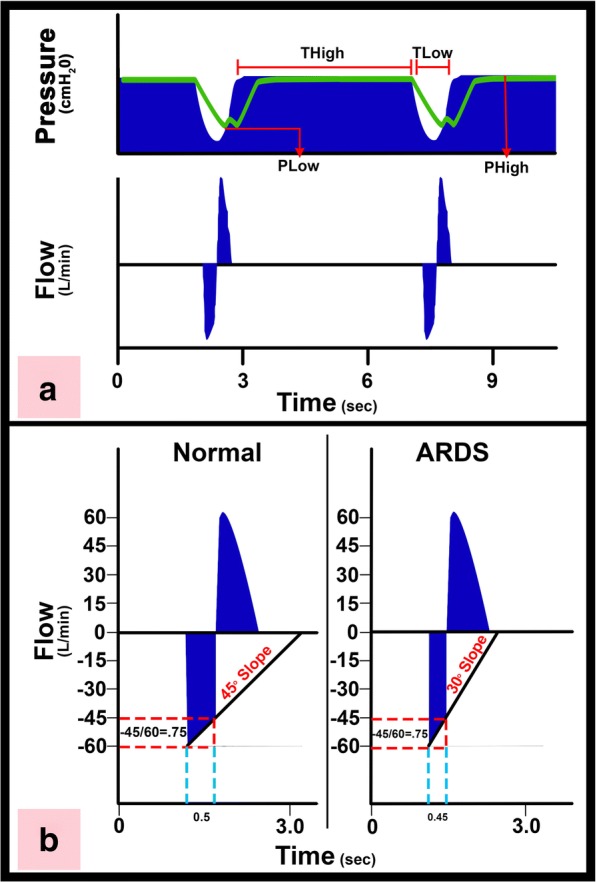
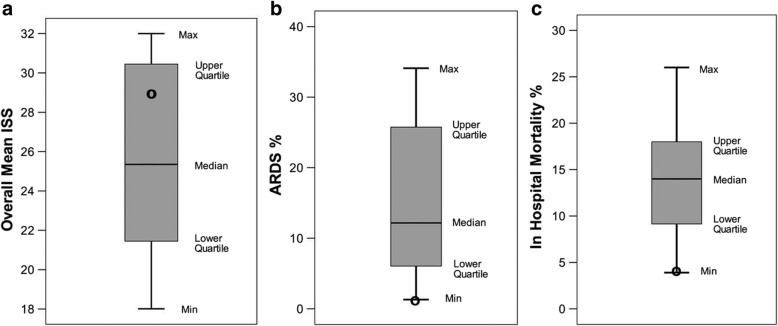
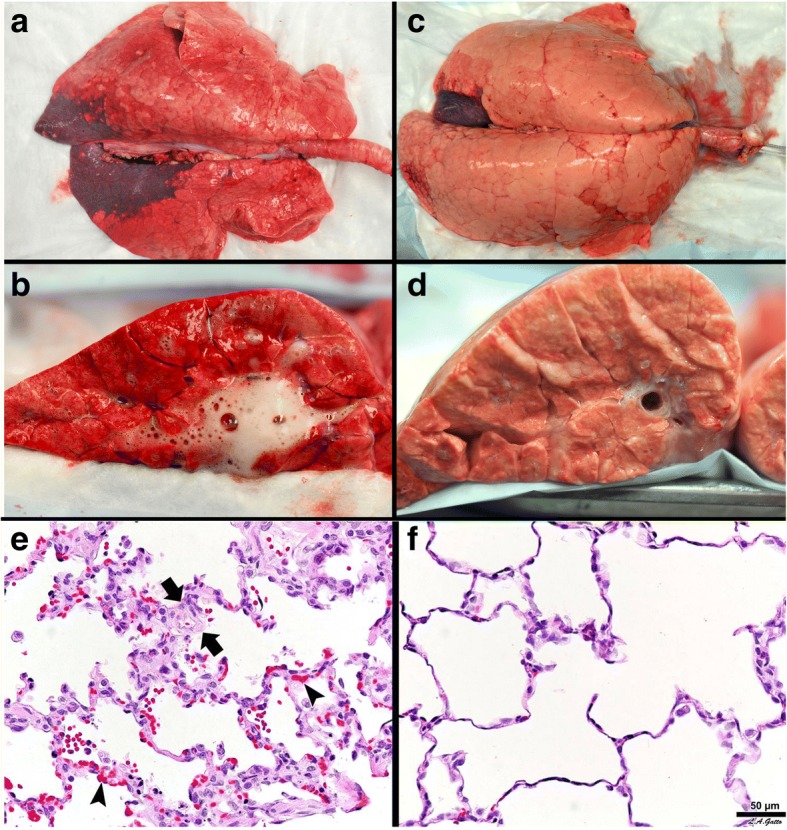
The pathophysiology of acute respiratory distress syndrome (ARDS) results in heterogeneous lung collapse, edema-flooded airways and unstable alveoli. These pathologic alterations in alveolar mechanics (i.e. dynamic change in alveolar size and shape with each breath) predispose the lung to secondary ventilator-induced lung injury (VILI). It is our viewpoint that the acutely injured lung can be recruited and stabilized with a mechanical breath until it heals, much like casting a broken bone until it mends. If the lung can be "casted" with a mechanical breath, VILI could be prevented and ARDS incidence significantly reduced.
Keywords: Acute lung injury; Injurious mechanical ventilation; TCAV protocol.
Conflict of interest statement
Ethics approval and consent to participate
Not applicable.
Competing interests
PLA, GFN, MKS, and NMH have presented and received honoraria and/or travel reimbursement at event(s) sponsored by Dräger Medical Systems, Inc., outside of the published work. PLA, GFN, and NMH have lectured for Intensive Care Online Network, Inc. (ICON). NMH is the founder of ICON, of which PLA is an employee. NMH holds patents on a method of initiating, managing, and/or weaning airway pressure release ventilation, as well as controlling a ventilator in accordance with the same, but these patents are not commercialized, licensed, or royalty-producing. The authors maintain that industry had no role in the design and conduct of the study; the collection, management, analysis, or interpretation of the data; or the preparation, review, or approval of the manuscript.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
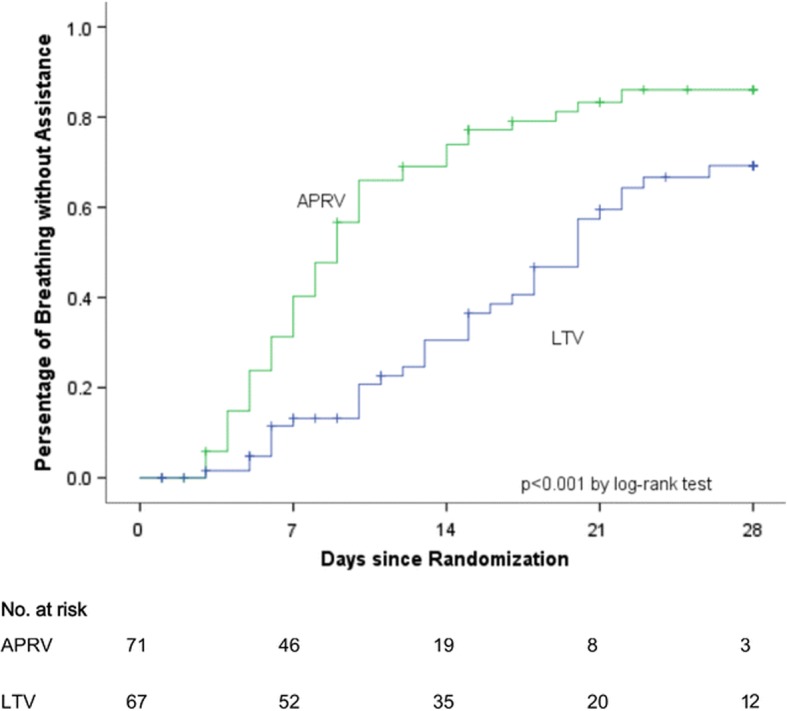
References
-
- Comroe JH. Physiology of respiration; an introductory text. 2. Chicago: Year Book Medical Publishers; 1974.
-
- Kollisch-Singule M, Emr B, Jain SV, Andrews P, Satalin J, Liu J, Porcellio E, Kenyon V, Wang G, Marx W, et al. The effects of airway pressure release ventilation on respiratory mechanics in extrapulmonary lung injury. Intensive Care Med Exp. 2015;3:35. doi: 10.1186/s40635-015-0071-0. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
