

A Rare De Novo RAI1 Gene Mutation Affecting BDNF-Enhancer-Driven Transcription Activity Associated with Autism and Atypical Smith-Magenis Syndrome Presentation
- PMID: 29794985
- PMCID: PMC6023015
- DOI: 10.3390/biology7020031
A Rare De Novo RAI1 Gene Mutation Affecting BDNF-Enhancer-Driven Transcription Activity Associated with Autism and Atypical Smith-Magenis Syndrome Presentation
Abstract
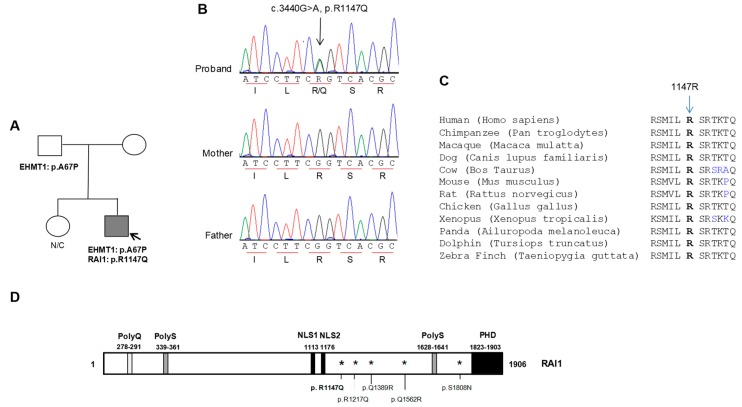
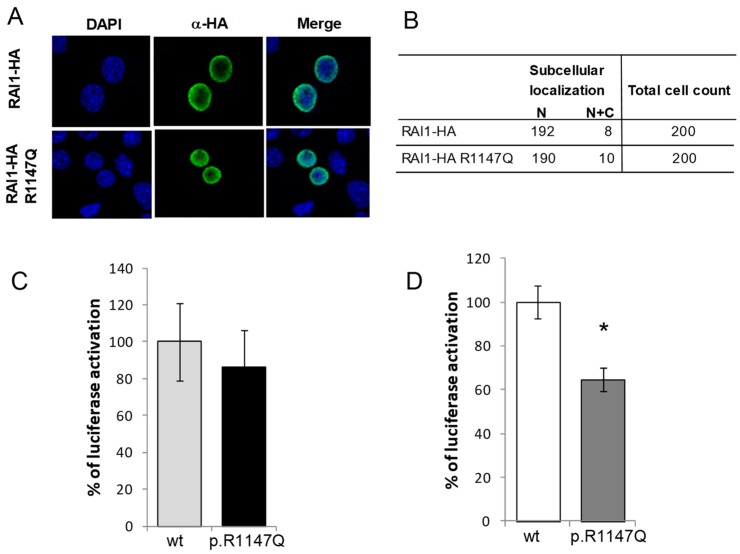
Deletions and mutations involving the Retinoic Acid Induced 1 (RAI1) gene at 17p11.2 cause Smith-Magenis syndrome (SMS). Here we report a patient with autism as the main clinical presentation, with some SMS-like features and a rare de novo RAI1 gene mutation, c.3440G > A (p.R1147Q). We functionally characterized the RAI1 p.R1147Q mutant protein. The mutation, located near the nuclear localization signal, had no effect on the subcellular localization of the mutant protein. However, similar to previously reported RAI1 missense mutations in SMS patients, the RAI1 p.R1147Q mutant protein showed a significant deficiency in activating in vivo transcription of a reporter gene driven by a BDNF (brain-derived neurotrophic factor) intronic enhancer. In addition, expression of other genes associated with neurobehavioral abnormalities and/or neurodevelopmental disorders were found to be altered in this patient. These results suggest a likely contribution of RAI1, either alone or in combination of other factors, to social behavior and reinforce the RAI1 gene as a candidate gene in patients with autistic manifestations or social behavioral abnormalities.
Keywords: RAI1; Smith-Magenis syndrome; autism spectrum disorder; mutation; neurodevelopmental disorder.
Conflict of interest statement
The Molecular and Cytogenetic Diagnostic Laboratories at the Greenwood Genetic Center receive revenue for Next Generation Sequencing panels, microarrays, and various other genetic tests.
Figures
References
-
- Greenberg F., Guzzetta V., Montes de Oca-Luna R., Magenis R.E., Smith A.C., Richter S.F., Kondo I., Dobyns W.B., Patel P.I., Lupski J.R. Molecular analysis of the Smith-Magenis syndrome: A possible contiguous-gene syndrome associated with del(17)(p11.2) Am. J. Hum. Genet. 1999;49:1207–1218. - PMC - PubMed
-
- Smith A.C.M., Magenis R.E., Elsea S.H. Overview of Smith-Magenis Syndrome. J. Assoc. Genet. Technol. 2005;31:163–167. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
