

Towards a complete map of the human long non-coding RNA transcriptome
- PMID: 29795125
- PMCID: PMC6451964
- DOI: 10.1038/s41576-018-0017-y
Towards a complete map of the human long non-coding RNA transcriptome
Abstract
Gene maps, or annotations, enable us to navigate the functional landscape of our genome. They are a resource upon which virtually all studies depend, from single-gene to genome-wide scales and from basic molecular biology to medical genetics. Yet present-day annotations suffer from trade-offs between quality and size, with serious but often unappreciated consequences for downstream studies. This is particularly true for long non-coding RNAs (lncRNAs), which are poorly characterized compared to protein-coding genes. Long-read sequencing technologies promise to improve current annotations, paving the way towards a complete annotation of lncRNAs expressed throughout a human lifetime.
Conflict of interest statement
Competing interests
The authors declare no competing interests.
Figures
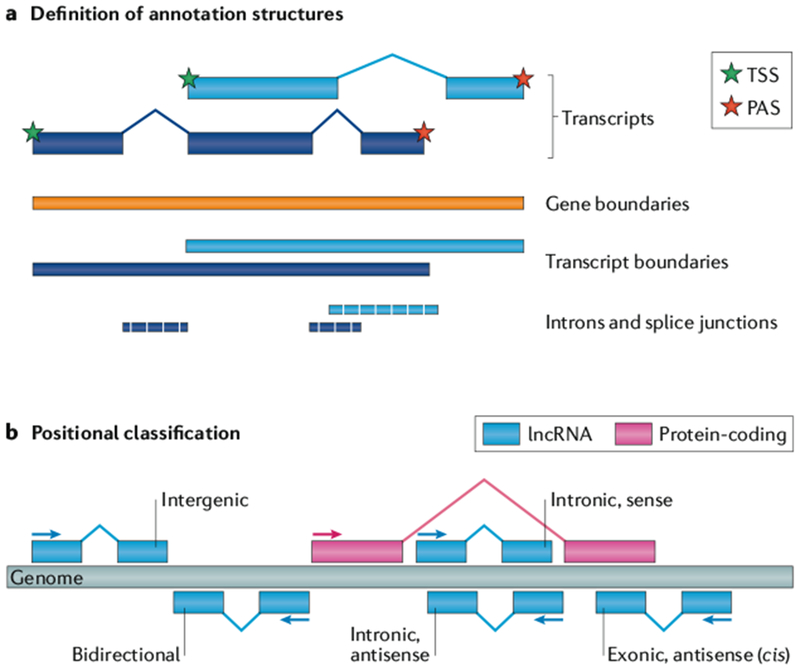
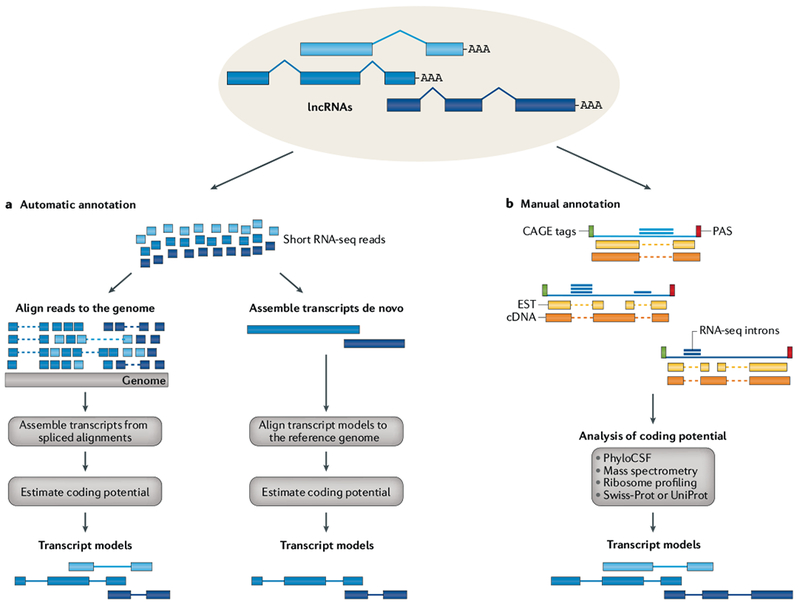
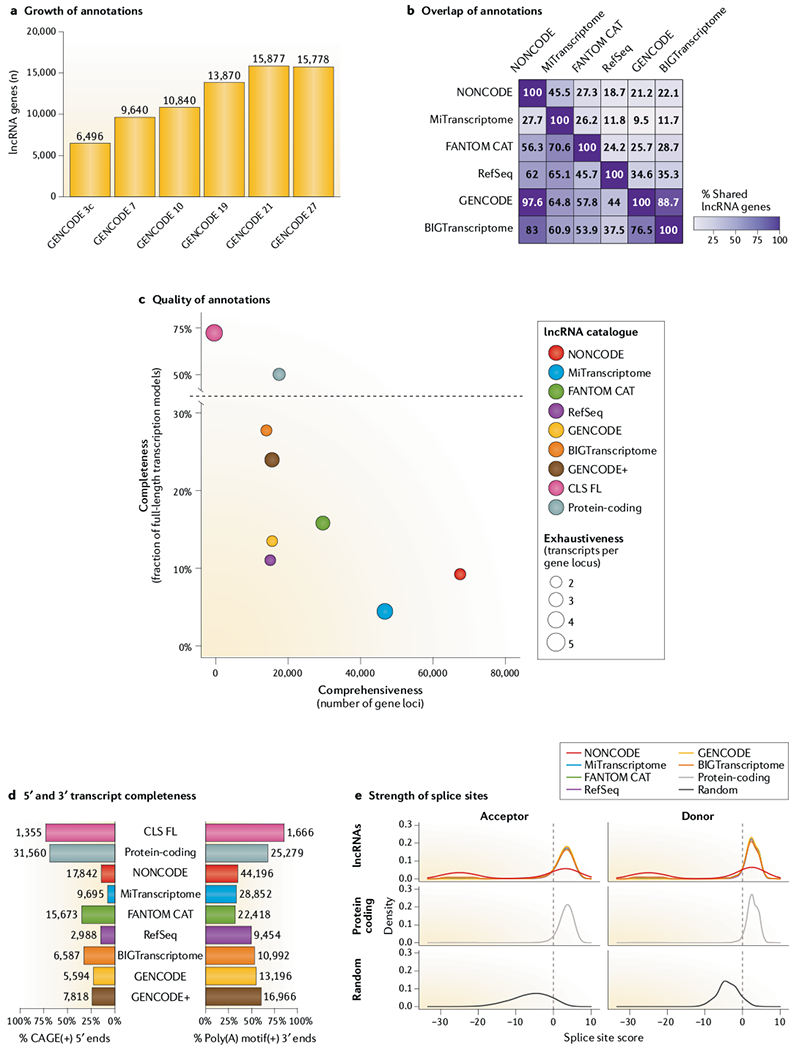
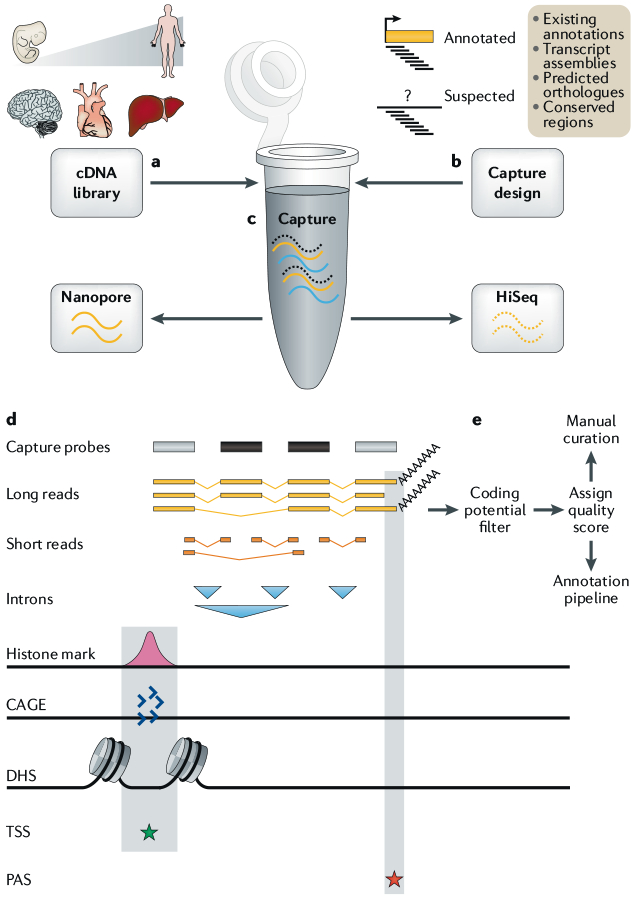
References
-
- Fang S et al. NONCODEV5: a comprehensive annotation database for long non-coding RNAs. Nucleic Acids Res. 46, D308–D314 (2018). - PMC - PubMed
-
This study presents the latest instalment of the long-running NONCODE annotation, which was amongst the first ncRNA annotations and currently represents the most extensive collection.
-
- Ponjavic J, Ponting CP & Lunter G Functionality or transcriptional noise? Evidence selection within long noncoding RNAs. Genome Res. 17, 556–565 (2007). - PMC - PubMed
-
This study initially demonstrated that lncRNA exons and promoters are under purifying evolutionary selection and hence provided strong evidence that, as a gene class, they are functional.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
