

NFκB mitigates the pathological effects of misfolded α1-antitrypsin by activating autophagy and an integrated program of proteostasis mechanisms
- PMID: 29795336
- PMCID: PMC6370812
- DOI: 10.1038/s41418-018-0130-7
NFκB mitigates the pathological effects of misfolded α1-antitrypsin by activating autophagy and an integrated program of proteostasis mechanisms
Abstract
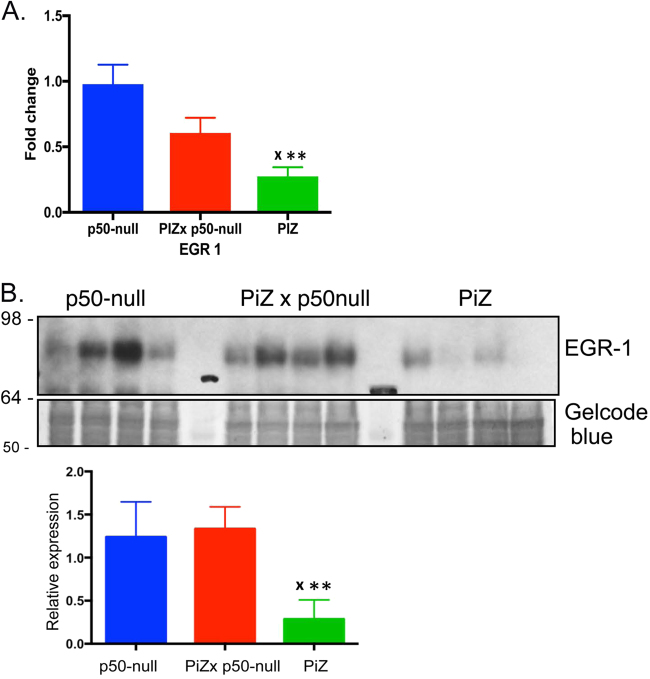
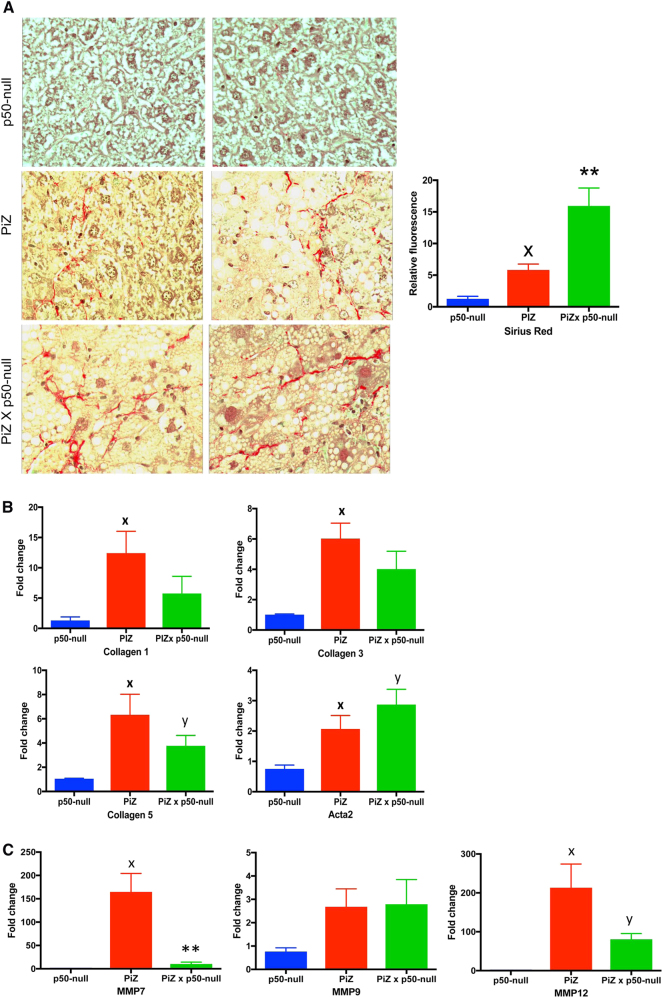
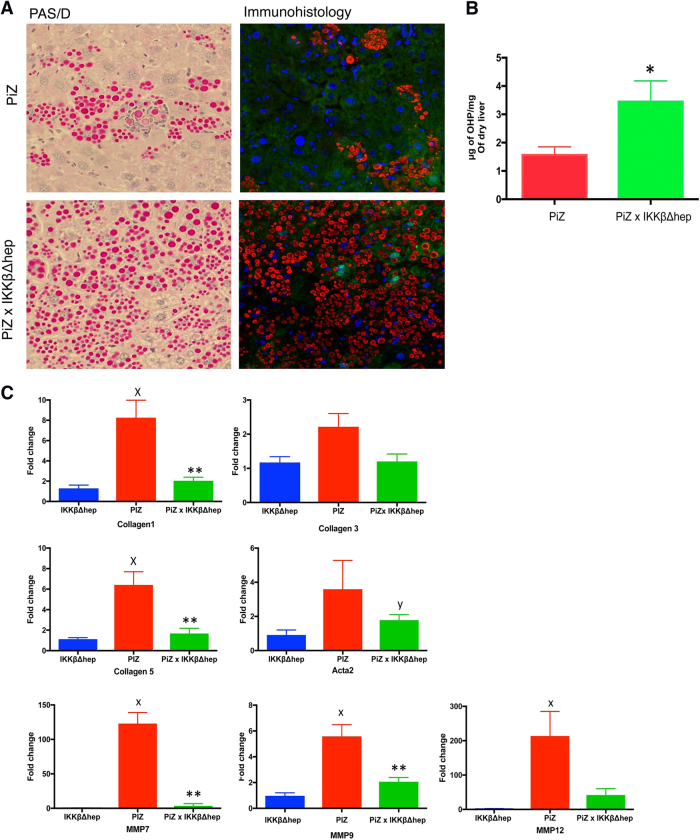
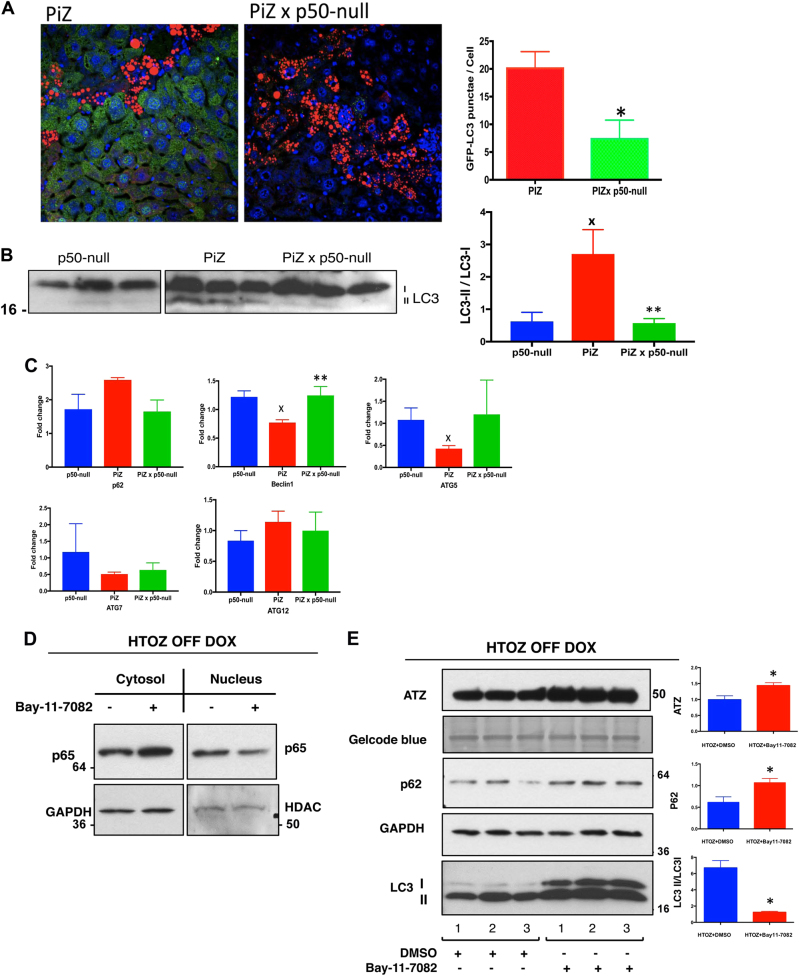
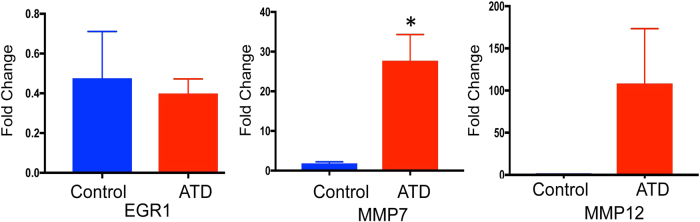
Intrahepatocytic accumulation of misfolded α1-antitrypsin Z variant (ATZ) is responsible for liver disease in some individuals with α1-antitrypsin deficiency (ATD), characterized by fibrosis/cirrhosis and predisposition to carcinogenesis. Previous results showing that accumulation of ATZ in model systems activates the NFκB signaling pathway have led us to hypothesize that downstream targets of NFκB are elements of a proteostasis response network for this type of proteinopathy. Here we show that only a subset of downstream targets within the NFκB transcriptomic repertoire are activated in model systems of this proteinopathy. Breeding of the PiZ mouse model of ATD to two different mouse models with NFκB deficiency led to greater intrahepatocytic accumulation of ATZ, more severe hepatic fibrosis, decreased autophagy and hyperproliferation of hepatocytes with massive ATZ inclusions. Specific downstream targets of NFκB could be implicated in each pathological effect. These results suggest a new role for NFκB signaling in which specific downstream targets of this pathway mediate an integrated program of proteostatic responses designed to mitigate the pathologic effects of proteinopathy, including autophagic disposal of misfolded protein, degradation of collagen and prevention of hyperproliferation.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures



References
-
- Hidvegi T, Mirnics K, Hale P, Ewing M, Beckett C, Perlmutter DH. Regulator of G signaling 16 is a marker for the distinct ER stress state associated with aggregated mutant α1-antitrypsin Z in the classical form of α1-antitrypsin deficiency. J Biol Chem. 2007;282:27769–80. doi: 10.1074/jbc.M704330200. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
