

Glutamate and GABA in autism spectrum disorder-a translational magnetic resonance spectroscopy study in man and rodent models
- PMID: 29802263
- PMCID: PMC5970172
- DOI: 10.1038/s41398-018-0155-1
Glutamate and GABA in autism spectrum disorder-a translational magnetic resonance spectroscopy study in man and rodent models
Abstract
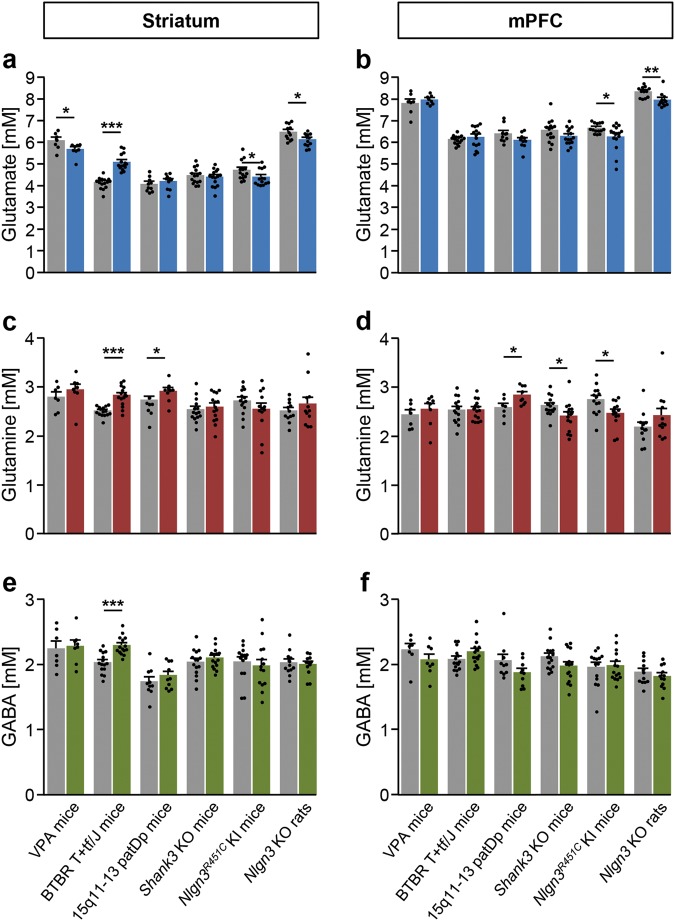
Autism spectrum disorder (ASD) is a pervasive neurodevelopmental syndrome with a high human and economic burden. The pathophysiology of ASD is largely unclear, thus hampering development of pharmacological treatments for the core symptoms of the disorder. Abnormalities in glutamate and GABA signaling have been hypothesized to underlie ASD symptoms, and may form a therapeutic target, but it is not known whether these abnormalities are recapitulated in humans with ASD, as well as in rodent models of the disorder. We used translational proton magnetic resonance spectroscopy ([1H]MRS) to compare glutamate and GABA levels in adult humans with ASD and in a panel of six diverse rodent ASD models, encompassing genetic and environmental etiologies. [1H]MRS was performed in the striatum and the medial prefrontal cortex, of the humans, mice, and rats in order to allow for direct cross-species comparisons in specific cortical and subcortical brain regions implicated in ASD. In humans with ASD, glutamate concentration was reduced in the striatum and this was correlated with the severity of social symptoms. GABA levels were not altered in either brain region. The reduction in striatal glutamate was recapitulated in mice prenatally exposed to valproate, and in mice and rats carrying Nlgn3 mutations, but not in rodent ASD models with other etiologies. Our findings suggest that glutamate/GABA abnormalities in the corticostriatal circuitry may be a key pathological mechanism in ASD; and may be linked to alterations in the neuroligin-neurexin signaling complex.
Conflict of interest statement
M.M.P., A.B., W.S., and B.K. received salaries for employment with F. Hoffmann-La. The remaining authors declare that they have no conflict of interest.
Figures

References
-
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders 5th edn (American Psychiatric Press, Arlington, TX, 2013).
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
