

Identification of rare de novo epigenetic variations in congenital disorders
- PMID: 29802345
- PMCID: PMC5970273
- DOI: 10.1038/s41467-018-04540-x
Identification of rare de novo epigenetic variations in congenital disorders
Abstract
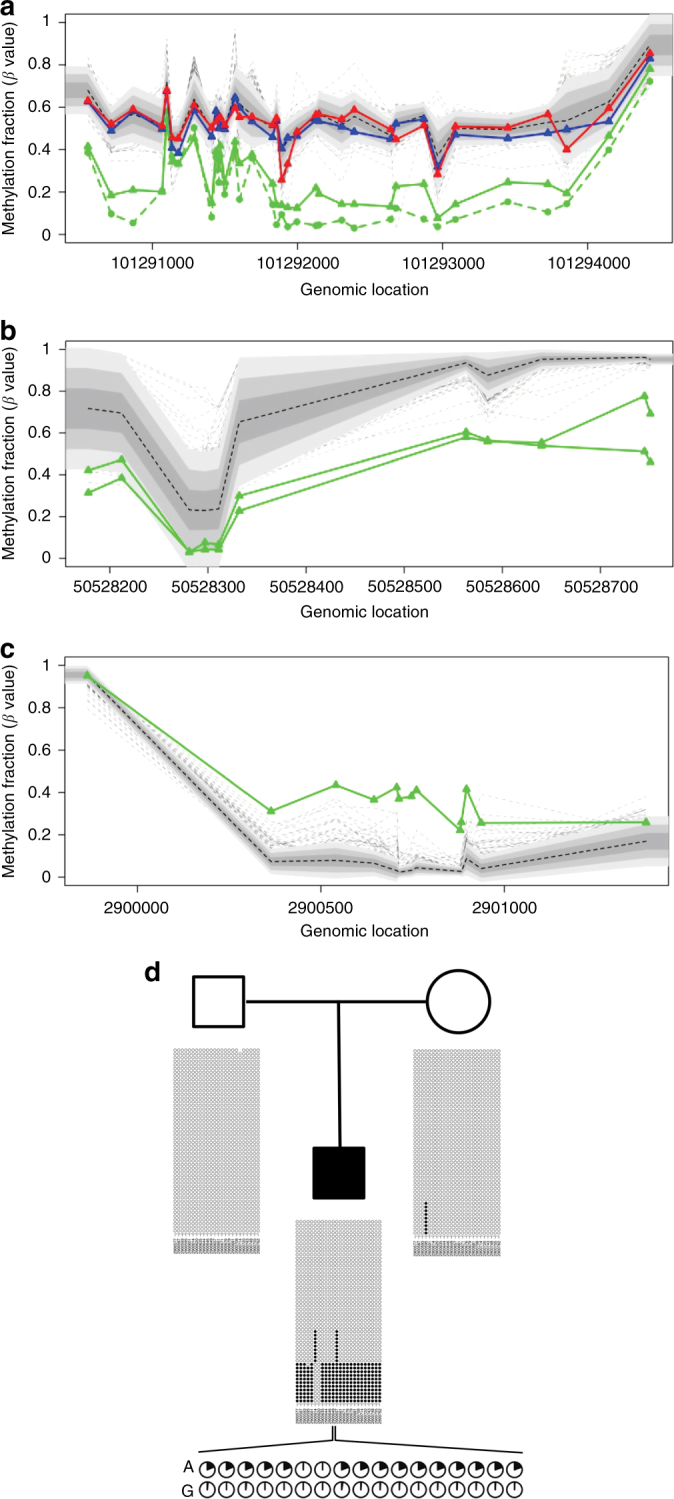
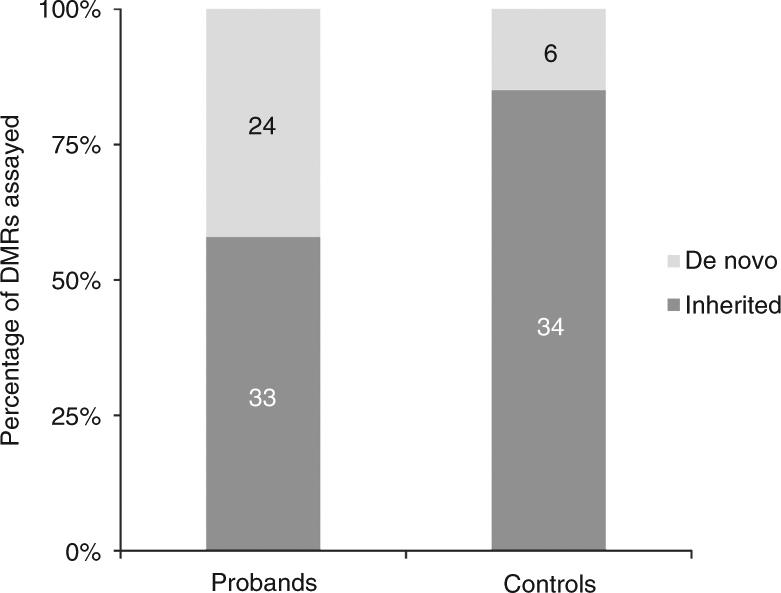
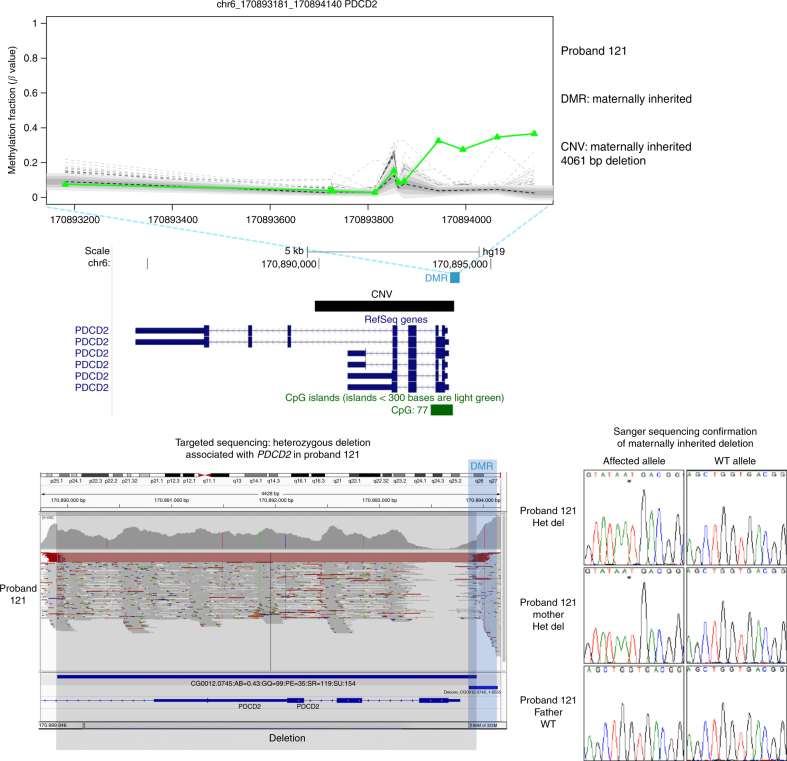
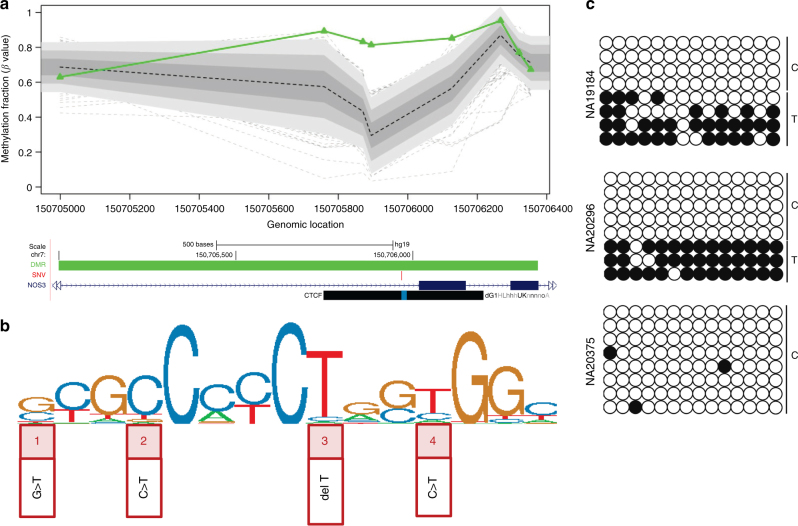
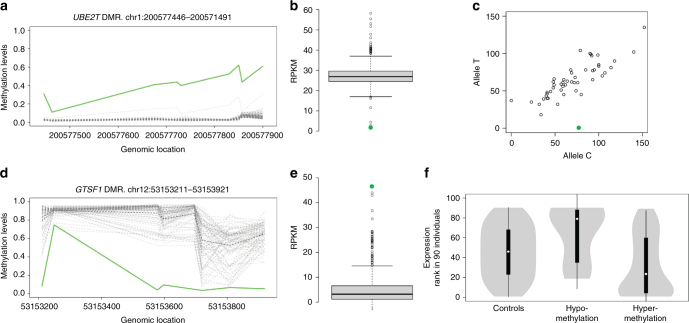
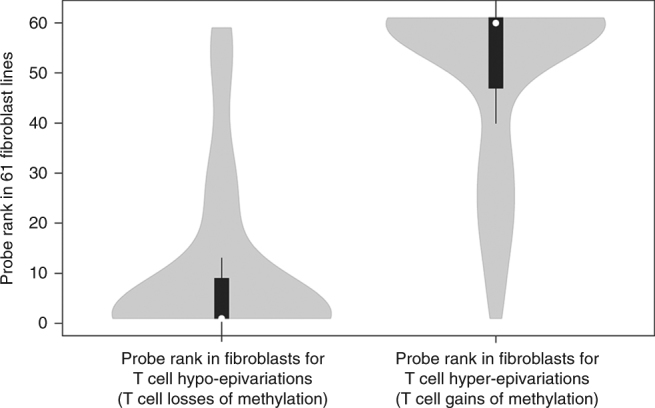
Certain human traits such as neurodevelopmental disorders (NDs) and congenital anomalies (CAs) are believed to be primarily genetic in origin. However, even after whole-genome sequencing (WGS), a substantial fraction of such disorders remain unexplained. We hypothesize that some cases of ND-CA are caused by aberrant DNA methylation leading to dysregulated genome function. Comparing DNA methylation profiles from 489 individuals with ND-CAs against 1534 controls, we identify epivariations as a frequent occurrence in the human genome. De novo epivariations are significantly enriched in cases, while RNAseq analysis shows that epivariations often have an impact on gene expression comparable to loss-of-function mutations. Additionally, we detect and replicate an enrichment of rare sequence mutations overlapping CTCF binding sites close to epivariations, providing a rationale for interpreting non-coding variation. We propose that epivariations contribute to the pathogenesis of some patients with unexplained ND-CAs, and as such likely have diagnostic relevance.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Horsthemke B. Epimutations in human disease. Curr. Top. Microbiol. Immunol. 2006;310:45–59. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
