

Secondary IgA nephropathy
- PMID: 29804660
- PMCID: PMC6981247
- DOI: 10.1016/j.kint.2018.02.030
Secondary IgA nephropathy
Abstract
IgA nephropathy is the most common primary glomerulonephritis worldwide. Its frequent coexistence with inflammatory, infectious, or malignant processes raises the possibility of a pathologic rather than coincidental association. Major strides have been made to elucidate the underlying pathophysiologic events that culminate in the development of primary IgA nephropathy. Whether secondary forms of the disease share common pathways triggered by underlying disorders or different mechanisms leading to similar pathologic findings remains to be determined. In this article we describe the most frequent etiologies for secondary IgA nephropathy and review the available literature for the pathophysiology.
Keywords: autoimmune diseases; glomerulonephritis; inflammatory bowel disease; liver disease; post-infectious glomerulonephritis.
Copyright © 2018 International Society of Nephrology. Published by Elsevier Inc. All rights reserved.
Figures
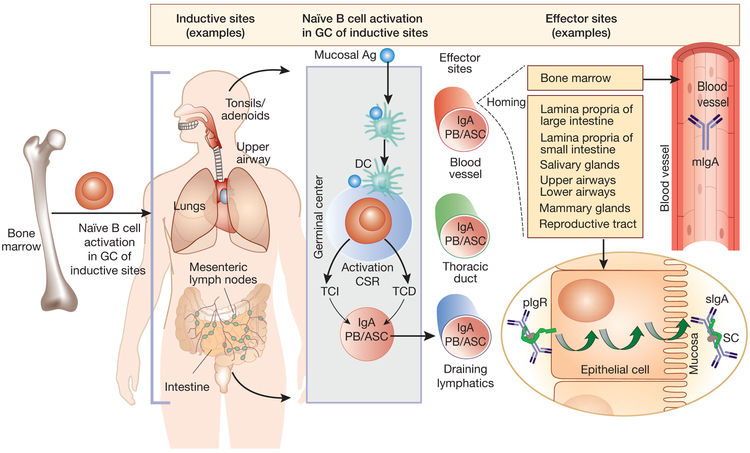
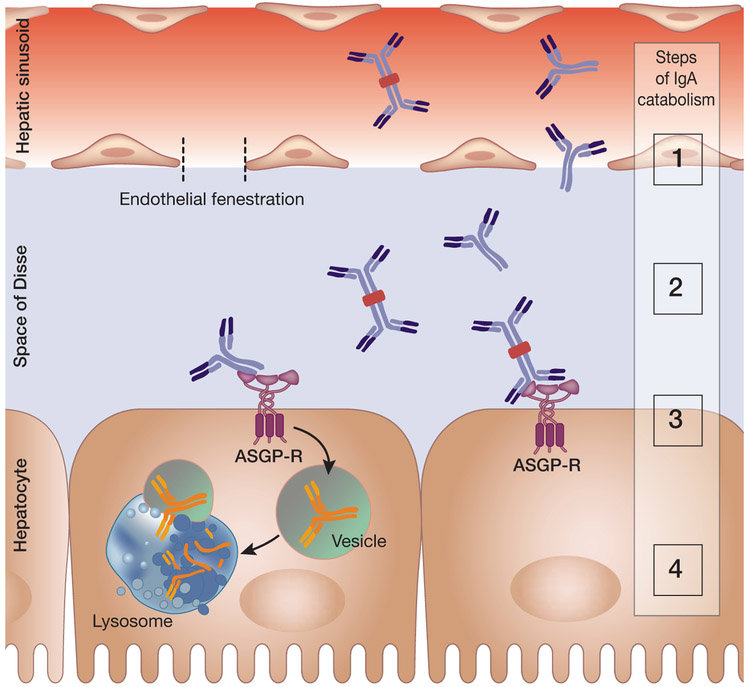
References
-
- Wyatt RJ, Julian BA. IgA nephropathy. N Engl J Med 2013; 368: 2402–2414. - PubMed
-
- Hiki Y, Odani H, Takahashi M, et al. Mass spectrometry proves under-O-glycosylation of glomerular IgA1 in IgA nephropathy. Kidney Int 2001; 59: 1077–1085. - PubMed
-
- Buren M, Yamashita M, Suzuki Y, et al. Altered expression of lymphocyte homing chemokines in the pathogenesis of IgA nephropathy. Contrib Nephrol 2007; 157: 50–55. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
