

Changes of signal transductivity and robustness of gene regulatory network in the carcinogenesis of leukemic subtypes via microarray sample data
- PMID: 29805763
- PMCID: PMC5955113
- DOI: 10.18632/oncotarget.25318
Changes of signal transductivity and robustness of gene regulatory network in the carcinogenesis of leukemic subtypes via microarray sample data
Abstract
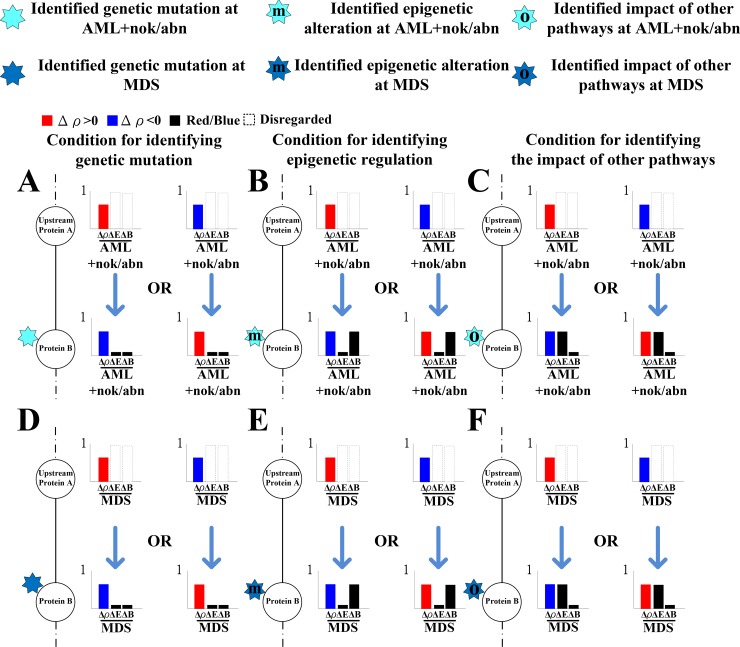
Mutation accumulation and epigenetic alterations in genes are important for carcinogenesis. Because leukemogenesis-related signal pathways have been investigated and microarray sample data have been produced in acute myeloid leukemia (AML), myelodysplastic syndromes (MDS) and normal cells, systems analysis in coupling pathways becomes possible. Based on system modeling and identification, we could construct the coupling pathways and their associated gene regulatory networks using microarray sample data. By applying system theory to the estimated system model in coupling pathways, we can then obtain transductivity sensitivity, basal sensitivity and error sensitivity of each protein to identify the potential impact of genetic mutations, epigenetic alterations and the coupling of other pathways from the perspective of energy, respectively. By comparing the results in AML, MDS and normal cells, we investigated the potential critical genetic mutations and epigenetic alterations that activate or repress specific cellular functions to promote MDS or AML leukemogenesis. We suggested that epigenetic modification of β-catenin and signal integration of CSLs, AP-2α, STATs, c-Jun and β-catenin could contribute to cell proliferation at AML and MDS. Epigenetic regulation of ERK and genetic mutation of p53 could lead to the repressed apoptosis, cell cycle arrest and DNA repair in leukemic cells. Genetic mutation of JAK, epigenetic regulation of ERK, and signal integration of C/EBPα could result in the promotion of MDS cell differentiation. According to the results, we proposed three drugs, decitabine, genistein, and monorden for preventing AML leukemogenesis, while three drugs, decitabine, thalidomide, and geldanamycin, for preventing MDS leukemogenesis.
Keywords: basal sensitivity; error sensitivity; network robustness; transductivity; transductivity sensitivity.
Conflict of interest statement
CONFLICTS OF INTEREST The authors declare that they have no competing interests.
Figures









References
-
- Steelman LS, Abrams SL, Whelan J, Bertrand FE, Ludwig DE, Bäsecke J, Libra M, Stivala F, Milella M, Tafuri A, Lunghi P, Bonati A, Martelli AM, McCubrey JA. Contributions of the Raf/MEK/ERK, PI3K/PTEN/Akt/mTOR and Jak/STAT pathways to leukemia. Leukemia. 2008;22:686–707. https://doi.org/10.1038/leu.2008.26. - DOI - PubMed
-
- Takahashi S. Downstream molecular pathways of FLT3 in the pathogenesis of acute myeloid leukemia: biology and therapeutic implications. J Hematol Oncol. 2011;4:13. https://doi.org/10.1186/1756-8722-4-13. - DOI - PMC - PubMed
-
- Koschmieder SKU. Myeloid Leukemia - Basic Mechanisms of Leukemogenesis. InTech. 2011.
-
- Zhang R, Shah MV, Yang J, Nyland SB, Liu X, Yun JK, Albert R, Loughran TP., Jr Network model of survival signaling in large granular lymphocyte leukemia. Proc Natl Acad Sci USA. 2008;105:16308–13. https://doi.org/10.1073/pnas.0806447105. - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
