

Transcriptome-wide association studies accounting for colocalization using Egger regression
- PMID: 29808603
- PMCID: PMC6342197
- DOI: 10.1002/gepi.22131
Transcriptome-wide association studies accounting for colocalization using Egger regression
Abstract
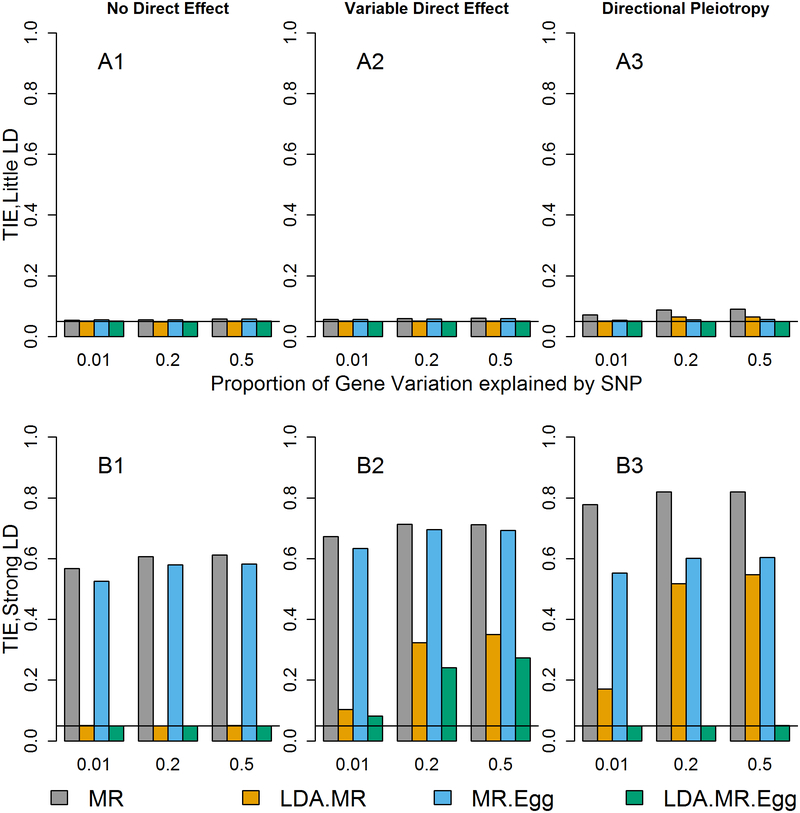
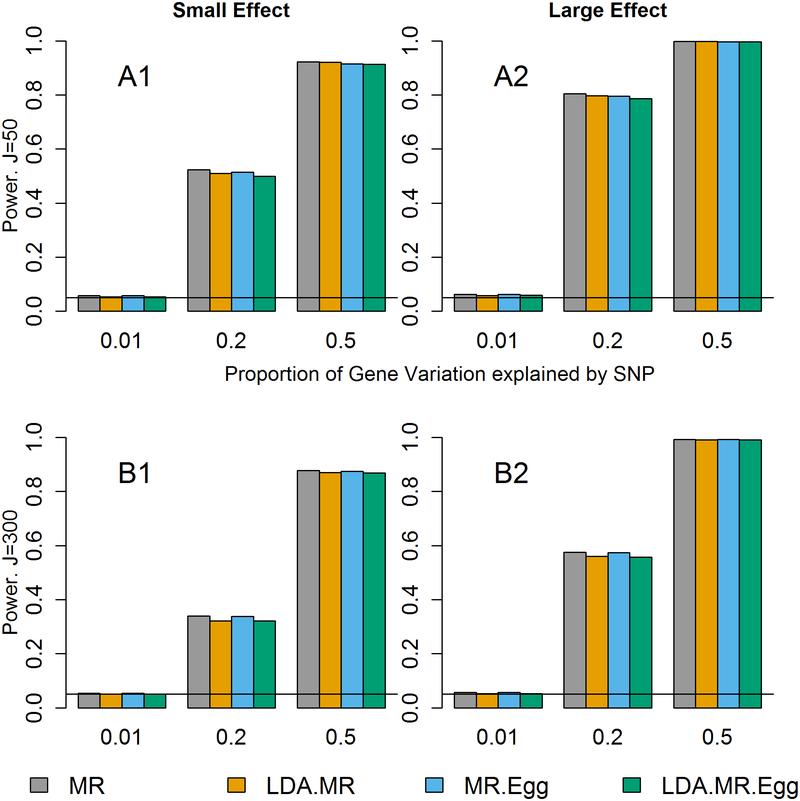
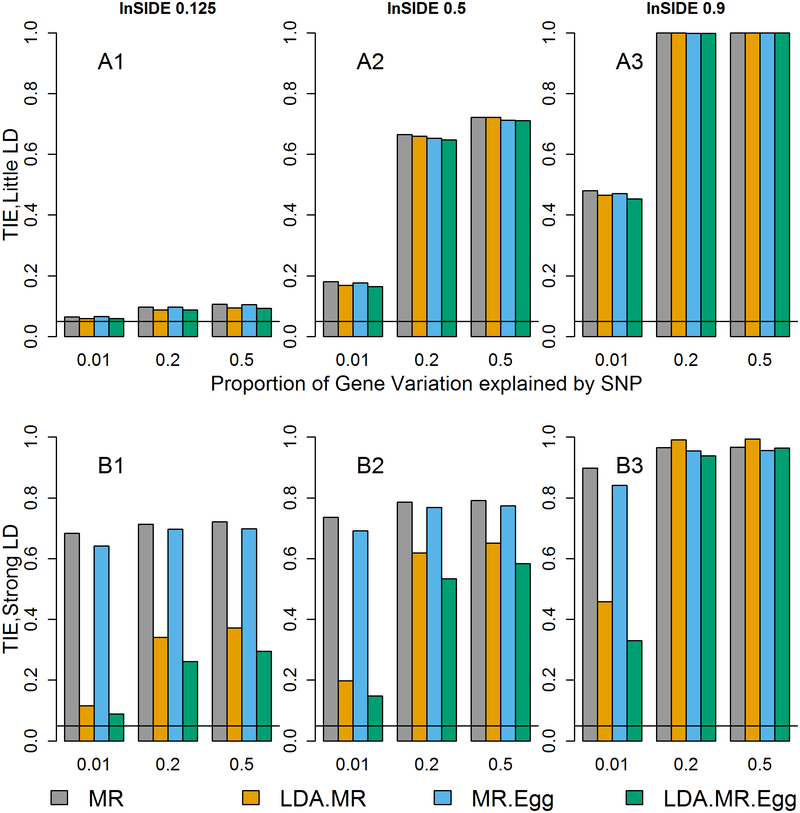
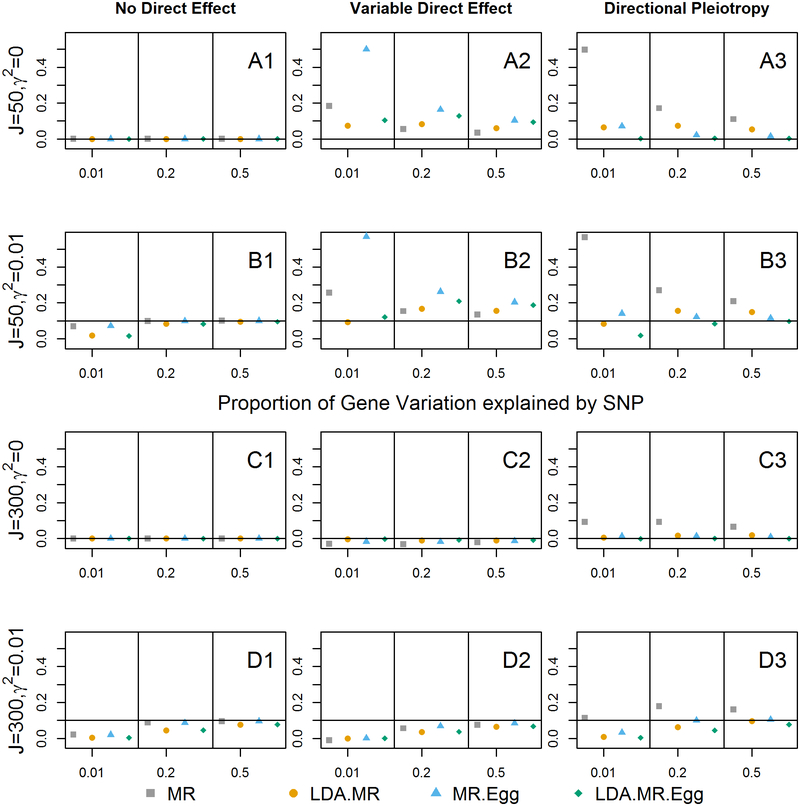
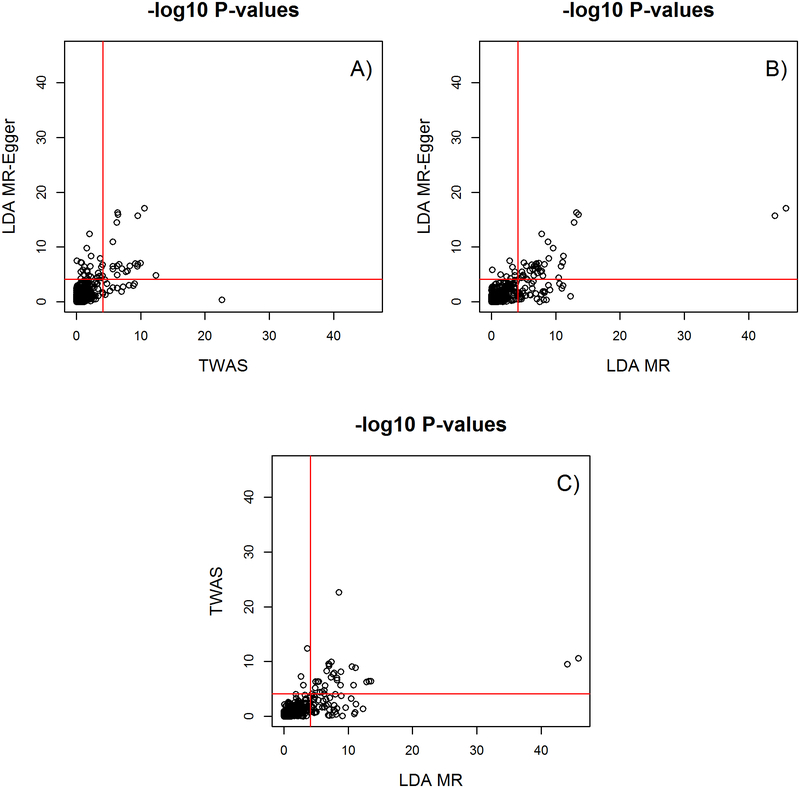
Integrating genome-wide association (GWAS) and expression quantitative trait locus (eQTL) data into transcriptome-wide association studies (TWAS) based on predicted expression can boost power to detect novel disease loci or pinpoint the susceptibility gene at a known disease locus. However, it is often the case that multiple eQTL genes colocalize at disease loci, making the identification of the true susceptibility gene challenging, due to confounding through linkage disequilibrium (LD). To distinguish between true susceptibility genes (where the genetic effect on phenotype is mediated through expression) and colocalization due to LD, we examine an extension of the Mendelian randomization (MR) egger regression method that allows for LD while only requiring summary association data for both GWAS and eQTL. We derive the standard TWAS approach in the context of MR and show in simulations that the standard TWAS does not control type I error for causal gene identification when eQTLs have pleiotropic or LD-confounded effects on disease. In contrast, LD-aware MR-Egger (LDA MR-Egger) regression can control type I error in this case while attaining similar power as other methods in situations where these provide valid tests. However, when the direct effects of genetic variants on traits are correlated with the eQTL associations, all of the methods we examined including LDA MR-Egger regression can have inflated type I error. We illustrate these methods by integrating gene expression within a recent large-scale breast cancer GWAS to provide guidance on susceptibility gene identification.
Keywords: Mendelian randomization; gene Expression; genome-wide association study; transciptome-wide association study.
© 2018 WILEY PERIODICALS, INC.
Figures
References
-
- Barbeira A, Shah KP, Torres JM, Wheeler HE, Torstenson ES, Edwards T, Garcia T, Bell GI, Nicolae D, Cox NJ and et al. 2016. MetaXcan: Summary Statistics Based Gene-Level Association Method Infers Accurate PrediXcan Results. bioRxiv.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
