

The Amyloid-β Oligomer Hypothesis: Beginning of the Third Decade
- PMID: 29843241
- PMCID: PMC6004937
- DOI: 10.3233/JAD-179941
The Amyloid-β Oligomer Hypothesis: Beginning of the Third Decade
Abstract
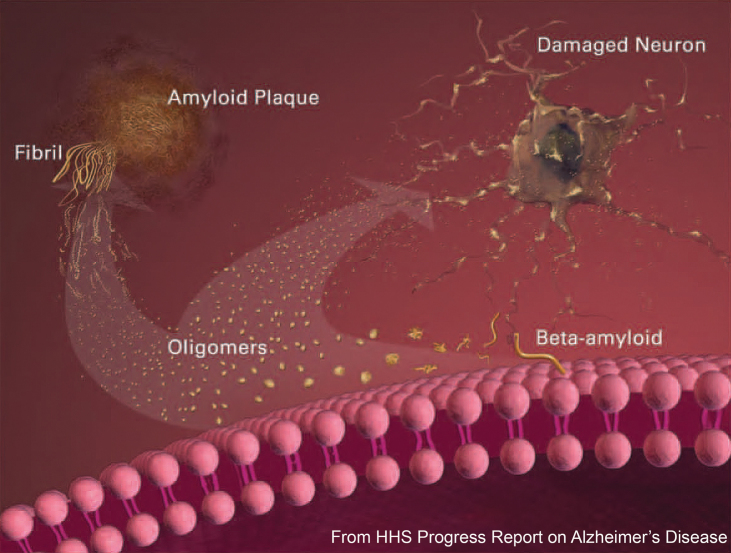
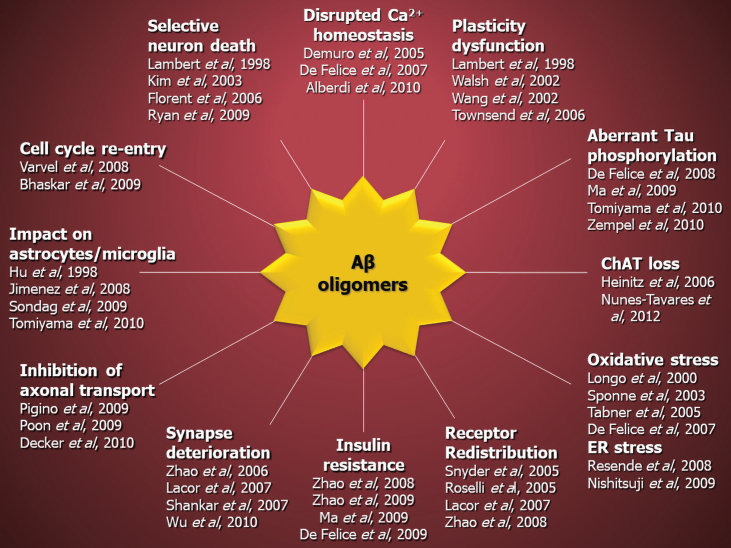
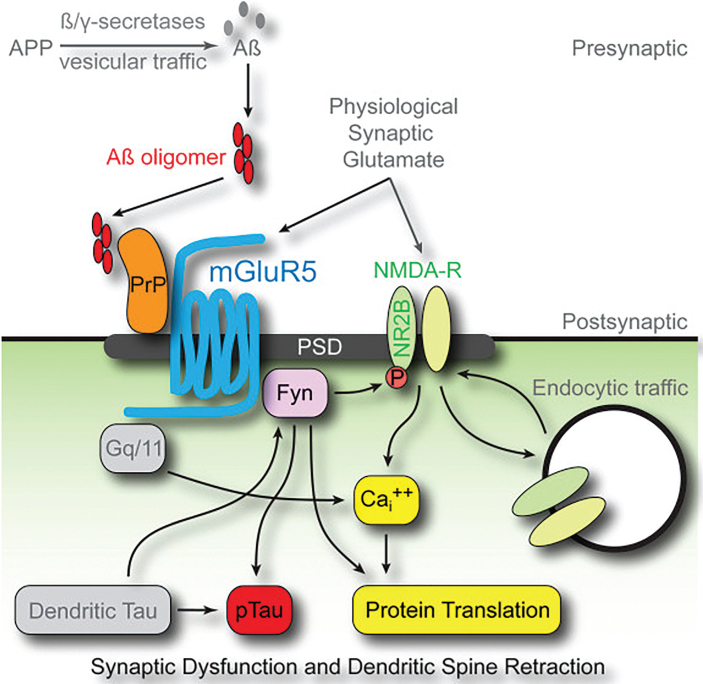
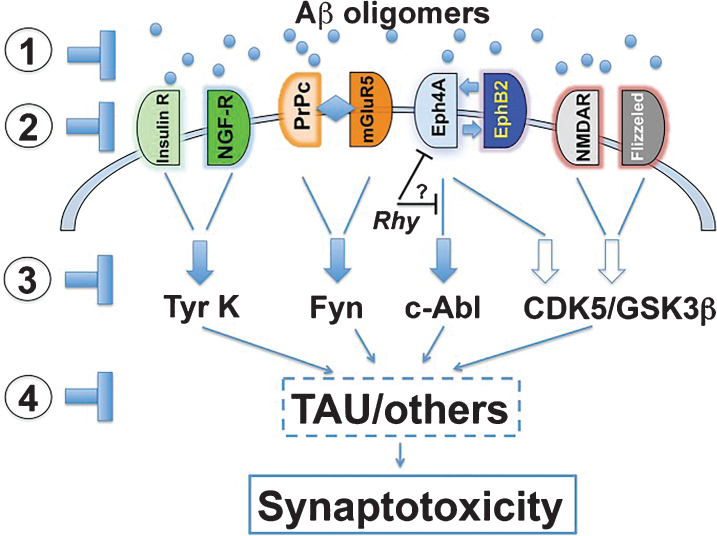
The amyloid-β oligomer (AβO) hypothesis was introduced in 1998. It proposed that the brain damage leading to Alzheimer's disease (AD) was instigated by soluble, ligand-like AβOs. This hypothesis was based on the discovery that fibril-free synthetic preparations of AβOs were potent CNS neurotoxins that rapidly inhibited long-term potentiation and, with time, caused selective nerve cell death (Lambert et al., 1998). The mechanism was attributed to disrupted signaling involving the tyrosine-protein kinase Fyn, mediated by an unknown toxin receptor. Over 4,000 articles concerning AβOs have been published since then, including more than 400 reviews. AβOs have been shown to accumulate in an AD-dependent manner in human and animal model brain tissue and, experimentally, to impair learning and memory and instigate major facets of AD neuropathology, including tau pathology, synapse deterioration and loss, inflammation, and oxidative damage. As reviewed by Hayden and Teplow in 2013, the AβO hypothesis "has all but supplanted the amyloid cascade." Despite the emerging understanding of the role played by AβOs in AD pathogenesis, AβOs have not yet received the clinical attention given to amyloid plaques, which have been at the core of major attempts at therapeutics and diagnostics but are no longer regarded as the most pathogenic form of Aβ. However, if the momentum of AβO research continues, particularly efforts to elucidate key aspects of structure, a clear path to a successful disease modifying therapy can be envisioned. Ensuring that lessons learned from recent, late-stage clinical failures are applied appropriately throughout therapeutic development will further enable the likelihood of a successful therapy in the near-term.
Keywords: Alzheimer’s disease; amyloid-β peptide; diagnostics; etiology; model systems; oligomers; prions; receptors; structure-function; tau; therapeutics.
Figures






References
-
- Frackowiak J, Zoltowska A, Wisniewski HM (1994) Non-fibrillar beta-amyloid protein is associated with smooth muscle cells of vessel walls in Alzheimer disease, J Neuropathol Exp Neurol 53, 637–645. - PubMed
-
- Oda T, Pasinetti GM, Osterburg HH, Anderson C, Johnson SA, Finch CE (1994) Purification and characterization of brain clusterin, Biochem Biophys Res Commun 204, 1131–1136. - PubMed
-
- Oda T, Wals P, Osterburg HH, Johnson SA, Pasinetti GM, Morgan TE, Rozovsky I, Stine WB, Snyder SW, Holzman TF, Krafft GA, Finch CE (1995) Clusterin (Apoj) alters the aggregation of amyloid beta-peptide (Aβ(1-42)) and forms slowly sedimenting Aβ complexes that cause oxidative stress, Exp Neurol 136, 22–31. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
