

Dominant-negative STAT5B mutations cause growth hormone insensitivity with short stature and mild immune dysregulation
- PMID: 29844444
- PMCID: PMC5974024
- DOI: 10.1038/s41467-018-04521-0
Dominant-negative STAT5B mutations cause growth hormone insensitivity with short stature and mild immune dysregulation
Abstract
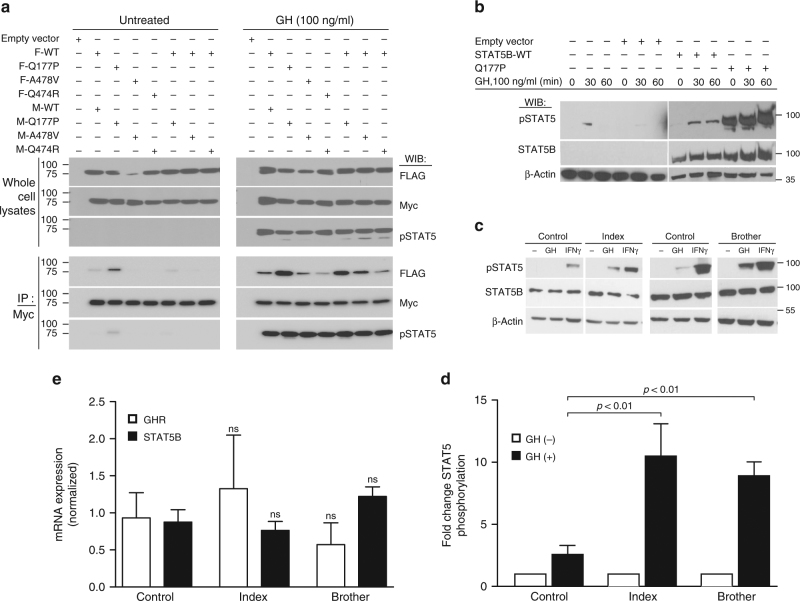
Growth hormone (GH) insensitivity syndrome (GHIS) is a rare clinical condition in which production of insulin-like growth factor 1 is blunted and, consequently, postnatal growth impaired. Autosomal-recessive mutations in signal transducer and activator of transcription (STAT5B), the key signal transducer for GH, cause severe GHIS with additional characteristics of immune and, often fatal, pulmonary complications. Here we report dominant-negative, inactivating STAT5B germline mutations in patients with growth failure, eczema, and elevated IgE but without severe immune and pulmonary problems. These STAT5B missense mutants are robustly tyrosine phosphorylated upon stimulation, but are unable to nuclear localize, or fail to bind canonical STAT5B DNA response elements. Importantly, each variant retains the ability to dimerize with wild-type STAT5B, disrupting the normal transcriptional functions of wild-type STAT5B. We conclude that these STAT5B variants exert dominant-negative effects through distinct pathomechanisms, manifesting in milder clinical GHIS with general sparing of the immune system.
Conflict of interest statement
R.G.R. consults for OPKO, Versartis, Ascendis, Genexine, Ammonite, Sandoz, Ferring, and NovoNordisk. The remaining authors declare no competing interests.
Figures


References
-
- Iida K, et al. Growth hormone (GH) insensitivity syndrome with high serum GH-binding protein levels caused by a heterozygous splice site mutation of the GH receptor gene producing a lack of intracellular domain. J. Clin. Endocrinol. Metab. 1998;83:531–537. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
