

Generation and validation of homozygous fluorescent knock-in cells using CRISPR-Cas9 genome editing
- PMID: 29844520
- PMCID: PMC6556379
- DOI: 10.1038/nprot.2018.042
Generation and validation of homozygous fluorescent knock-in cells using CRISPR-Cas9 genome editing
Abstract
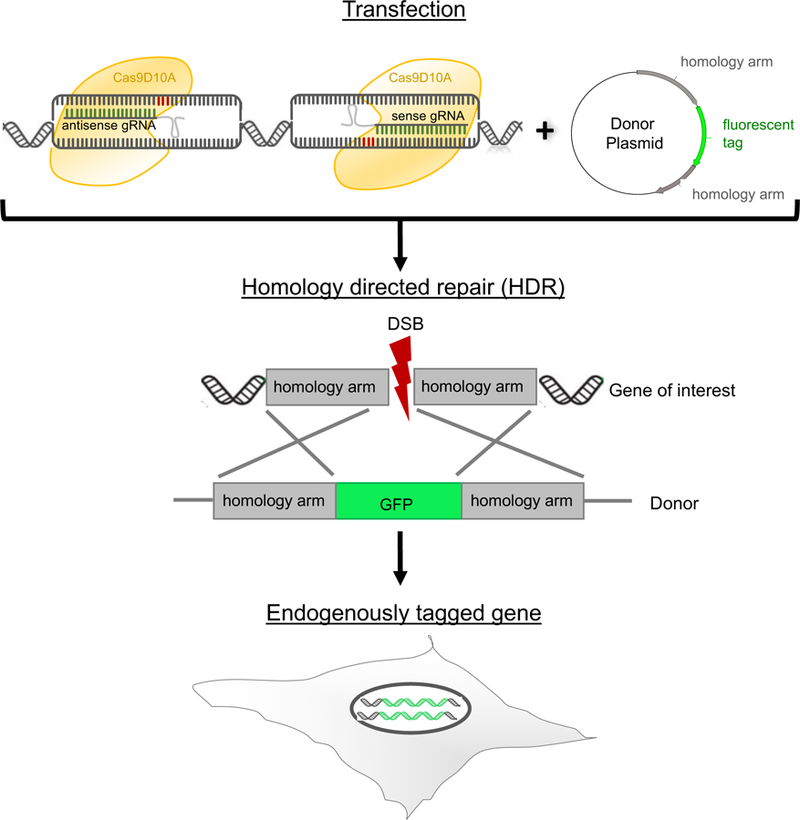
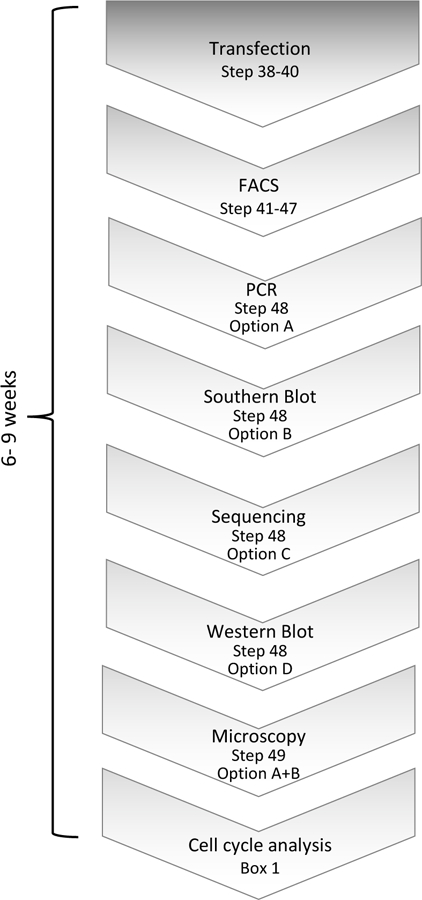
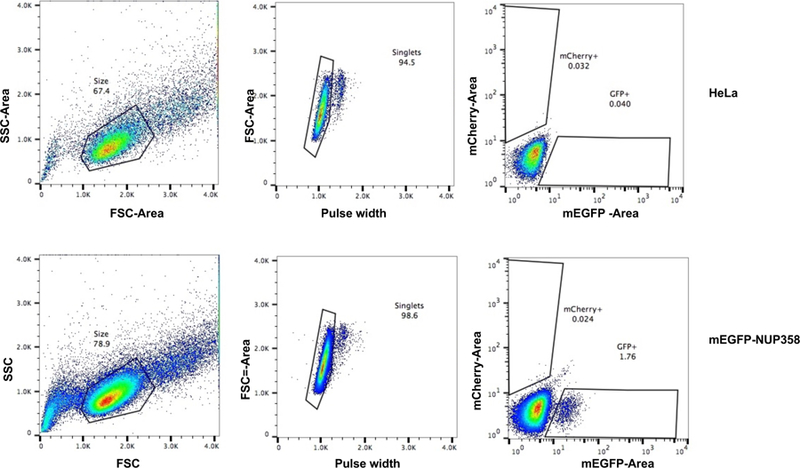
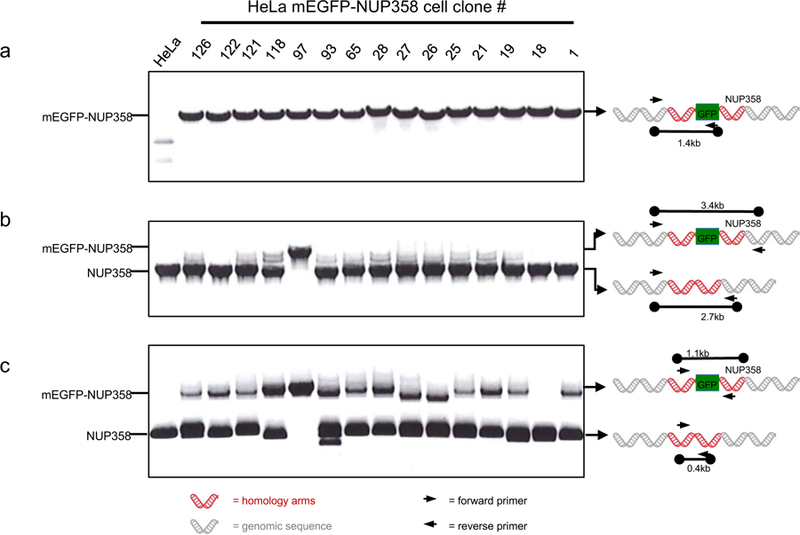
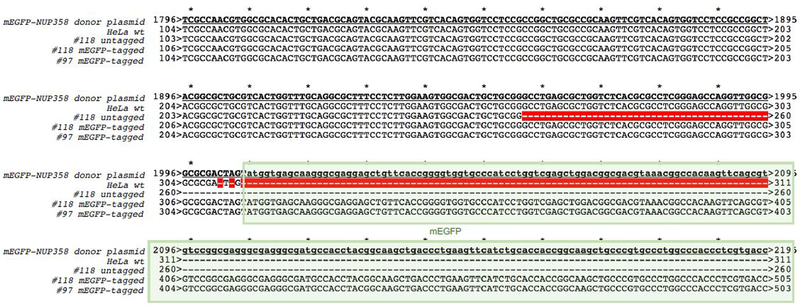
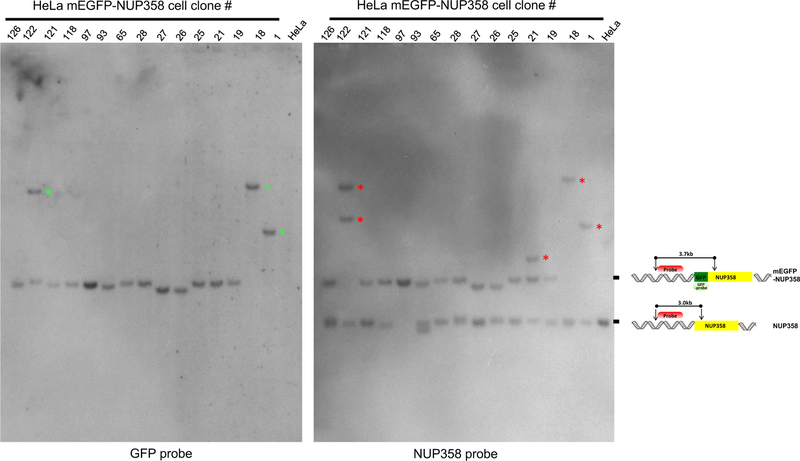
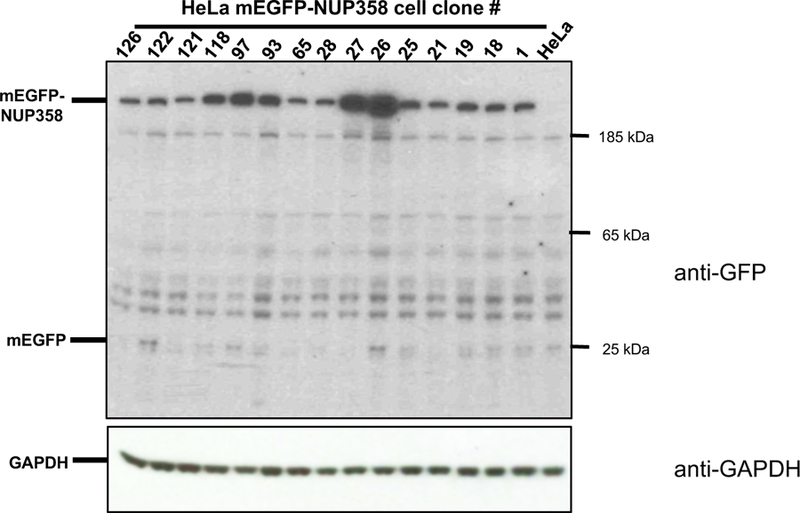
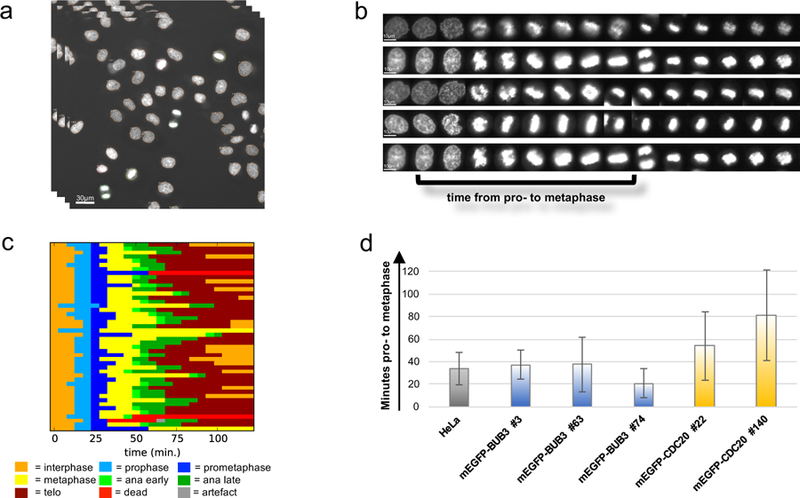
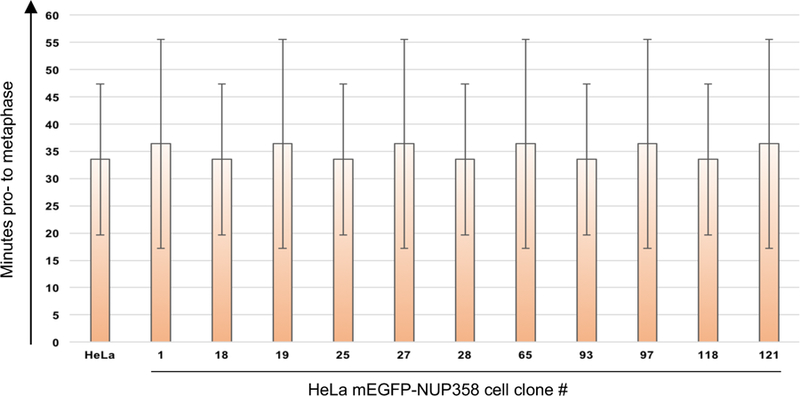
Gene tagging with fluorescent proteins is essential for investigations of the dynamic properties of cellular proteins. CRISPR-Cas9 technology is a powerful tool for inserting fluorescent markers into all alleles of the gene of interest (GOI) and allows functionality and physiological expression of the fusion protein. It is essential to evaluate such genome-edited cell lines carefully in order to preclude off-target effects caused by (i) incorrect insertion of the fluorescent protein, (ii) perturbation of the fusion protein by the fluorescent proteins or (iii) nonspecific genomic DNA damage by CRISPR-Cas9. In this protocol, we provide a step-by-step description of our systematic pipeline to generate and validate homozygous fluorescent knock-in cell lines.We have used the paired Cas9D10A nickase approach to efficiently insert tags into specific genomic loci via homology-directed repair (HDR) with minimal off-target effects. It is time-consuming and costly to perform whole-genome sequencing of each cell clone to check for spontaneous genetic variations occurring in mammalian cell lines. Therefore, we have developed an efficient validation pipeline of the generated cell lines consisting of junction PCR, Southern blotting analysis, Sanger sequencing, microscopy, western blotting analysis and live-cell imaging for cell-cycle dynamics. This protocol takes between 6 and 9 weeks. With this protocol, up to 70% of the targeted genes can be tagged homozygously with fluorescent proteins, thus resulting in physiological levels and phenotypically functional expression of the fusion proteins.
Conflict of interest statement
Competing interests
The authors declare that they have no competing financial interests.
Figures
References
-
- Trevino AE, et al. Genome Editing Using Cas9 Nickases. Methods Enzymol 546:161–74 (2014) - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
