

Determining the incidence of familiality in ALS: A study of temporal trends in Ireland from 1994 to 2016
- PMID: 29845113
- PMCID: PMC5961194
- DOI: 10.1212/NXG.0000000000000239
Determining the incidence of familiality in ALS: A study of temporal trends in Ireland from 1994 to 2016
Abstract
Objective: To assess temporal trends in familial amyotrophic lateral sclerosis (FALS) incidence rates in an Irish population and to determine factors influencing FALS ascertainment.
Methods: Population-based data collected over 23 years, using the Irish amyotrophic lateral sclerosis (ALS) register and DNA biobank, were analyzed and age-standardized rates of FALS and associated familial neuropsychiatric endophenotypes were identified.
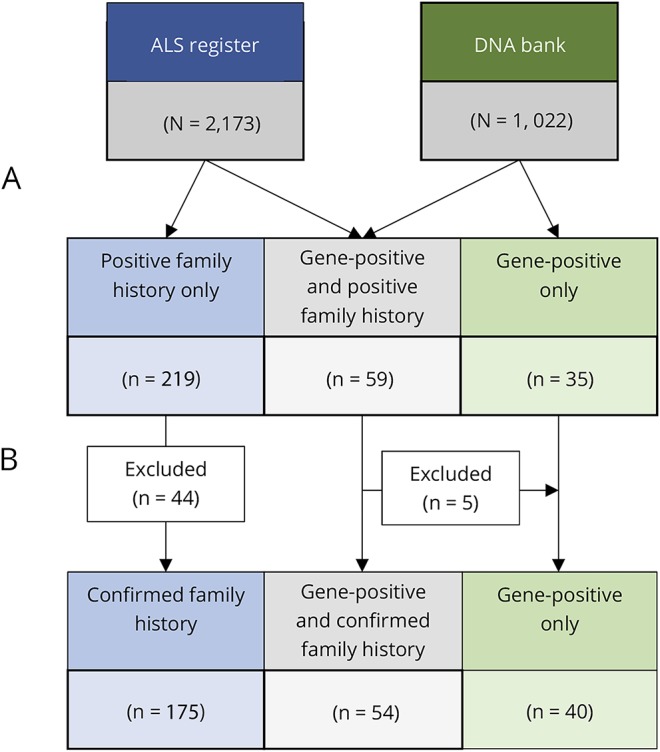
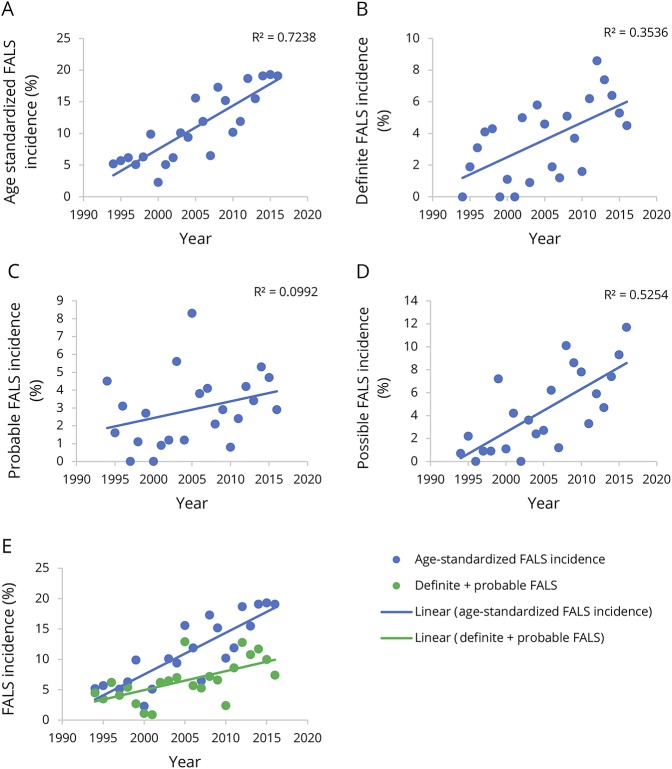
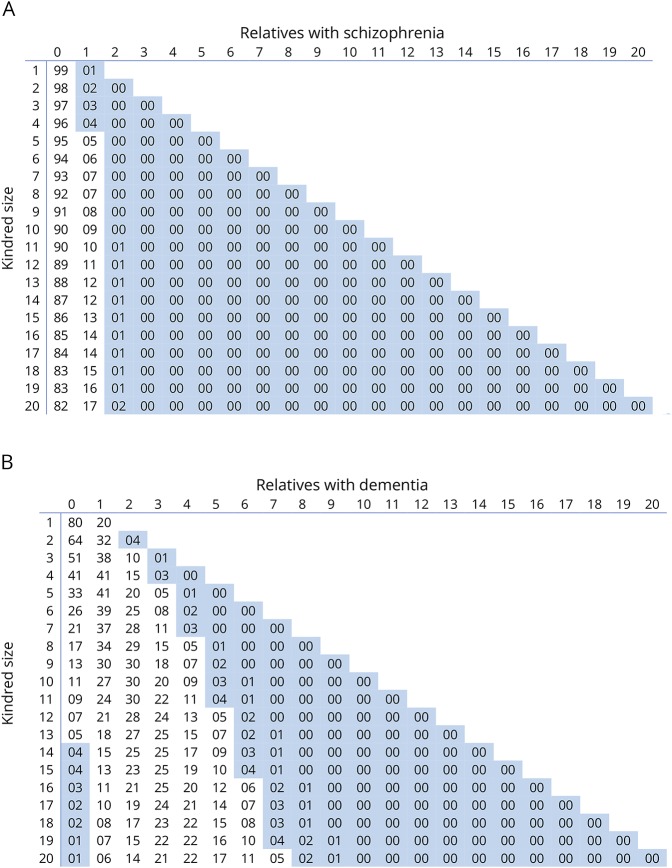
Results: Between 1994 and 2016, 269 patients with a family history of ALS from 197 unique families were included on the register. Using stringent diagnostic criteria for FALS, the mean age-standardized FALS incidence rate for the study period was 11.1% (95% confidence interval [CI], 8.8-13.4). The FALS incidence rate increased steadily from 5.2% in 1994 to 19.1% in 2016, an annual increase of 0.7% (95% CI, 0.5-0.9, p < 0.0001). Inclusion of the presence of neuropsychiatric endophenotypes within kindreds increased the FALS incidence rate to 30%. The incidence of FALS in newly diagnosed individuals from known families increased significantly with time, accounting for 50% of all FALS diagnoses by 2016. The mean annual rate of recategorization from "sporadic ALS" to "FALS" was 3% (95% CI, 2.6-3.8).
Conclusions: The true population-based rate of FALS is at least 20%. Inclusion of extended endophenotypes within kindreds increases the rate of FALS to 30%. Cross-sectional analysis of clinic-based cohorts and stringent definitions of FALS underestimate the true rate of familial disease. This has implications for genetic counseling and in the recognition of presymptomatic stages of ALS.
Figures

References
-
- Al-Chalabi A, van den Berg LH, Veldink J. Gene discovery in amyotrophic lateral sclerosis: implications for clinical management. Nat Rev Neurol 2017;13:96–104. - PubMed
-
- Jackson M, Al-Chalabi A, Enayat ZE, Chioza B, Leigh PN, Morrison KE. Copper/zinc superoxide dismutase 1 and sporadic amyotrophic lateral sclerosis: analysis of 155 cases and identification of a novel insertion mutation. Ann Neurol 1997;42:803–807. - PubMed
-
- Kabashi E, Valdmanis PN, Dion P, et al. . TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet 2008;40:572–574. - PubMed
-
- Hubers A, Just W, Rosenbohm A, et al. . De novo FUS mutations are the most frequent genetic cause in early-onset German ALS patients. Neurobiol Aging 2015;36:3117.e1–3117.e6. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous