

zUMIs - A fast and flexible pipeline to process RNA sequencing data with UMIs
- PMID: 29846586
- PMCID: PMC6007394
- DOI: 10.1093/gigascience/giy059
zUMIs - A fast and flexible pipeline to process RNA sequencing data with UMIs
Abstract
Background: Single-cell RNA-sequencing (scRNA-seq) experiments typically analyze hundreds or thousands of cells after amplification of the cDNA. The high throughput is made possible by the early introduction of sample-specific bar codes (BCs), and the amplification bias is alleviated by unique molecular identifiers (UMIs). Thus, the ideal analysis pipeline for scRNA-seq data needs to efficiently tabulate reads according to both BC and UMI.
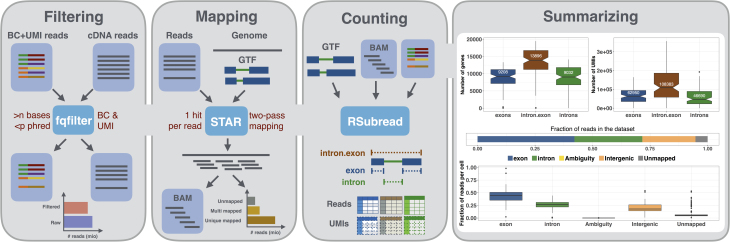
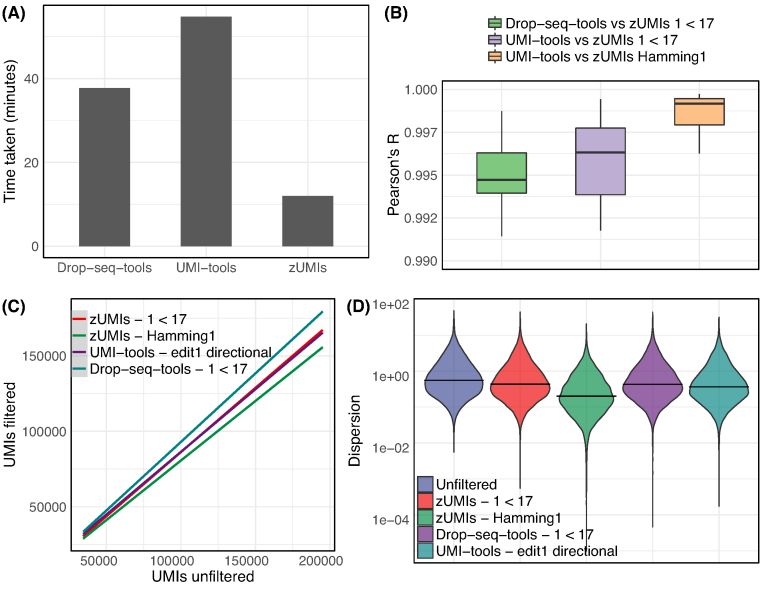
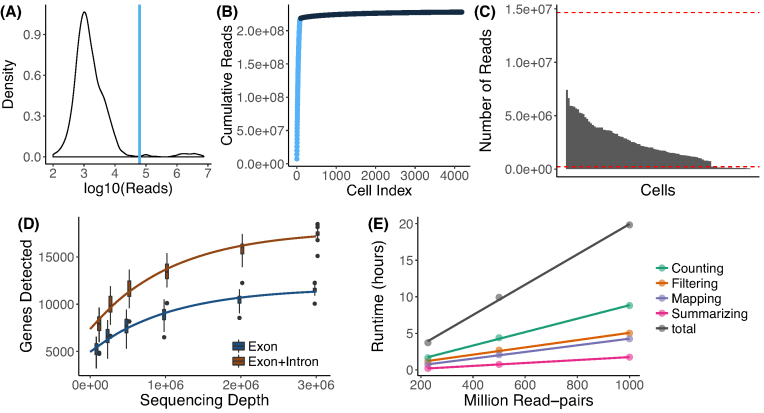
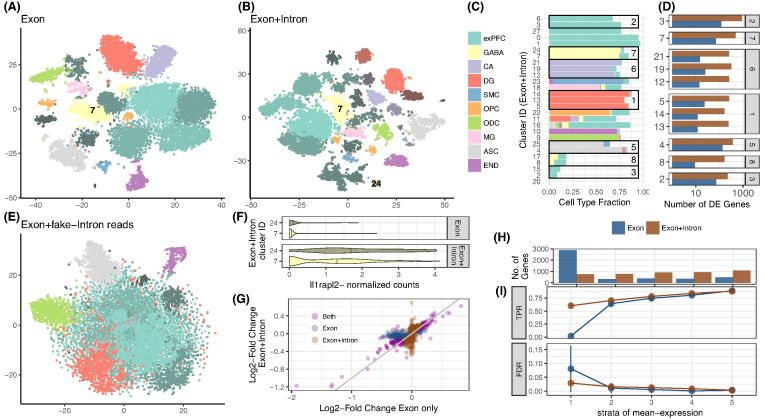
Findings: zUMIs is a pipeline that can handle both known and random BCs and also efficiently collapse UMIs, either just for exon mapping reads or for both exon and intron mapping reads. If BC annotation is missing, zUMIs can accurately detect intact cells from the distribution of sequencing reads. Another unique feature of zUMIs is the adaptive downsampling function that facilitates dealing with hugely varying library sizes but also allows the user to evaluate whether the library has been sequenced to saturation. To illustrate the utility of zUMIs, we analyzed a single-nucleus RNA-seq dataset and show that more than 35% of all reads map to introns. Also, we show that these intronic reads are informative about expression levels, significantly increasing the number of detected genes and improving the cluster resolution.
Conclusions: zUMIs flexibility makes if possible to accommodate data generated with any of the major scRNA-seq protocols that use BCs and UMIs and is the most feature-rich, fast, and user-friendly pipeline to process such scRNA-seq data.
Figures
References
-
- Sandberg R. Entering the era of single-cell transcriptomics in biology and medicine. Nat Methods. 2014;11(1):22–4. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
