

Mind the gap! The mitochondrial control region and its power as a phylogenetic marker in echinoids
- PMID: 29848319
- PMCID: PMC5977486
- DOI: 10.1186/s12862-018-1198-x
Mind the gap! The mitochondrial control region and its power as a phylogenetic marker in echinoids
Abstract
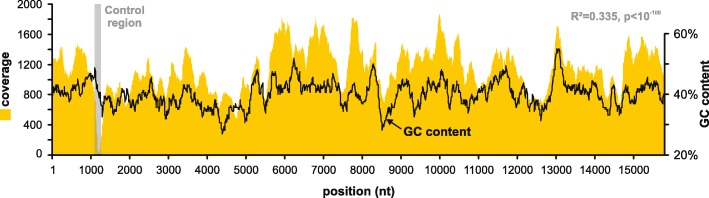
Background: In Metazoa, mitochondrial markers are the most commonly used targets for inferring species-level molecular phylogenies due to their extremely low rate of recombination, maternal inheritance, ease of use and fast substitution rate in comparison to nuclear DNA. The mitochondrial control region (CR) is the main non-coding area of the mitochondrial genome and contains the mitochondrial origin of replication and transcription. While sequences of the cytochrome oxidase subunit 1 (COI) and 16S rRNA genes are the prime mitochondrial markers in phylogenetic studies, the highly variable CR is typically ignored and not targeted in such analyses. However, the higher substitution rate of the CR can be harnessed to infer the phylogeny of closely related species, and the use of a non-coding region alleviates biases resulting from both directional and purifying selection. Additionally, complete mitochondrial genome assemblies utilizing next generation sequencing (NGS) data often show exceptionally low coverage at specific regions, including the CR. This can only be resolved by targeted sequencing of this region.
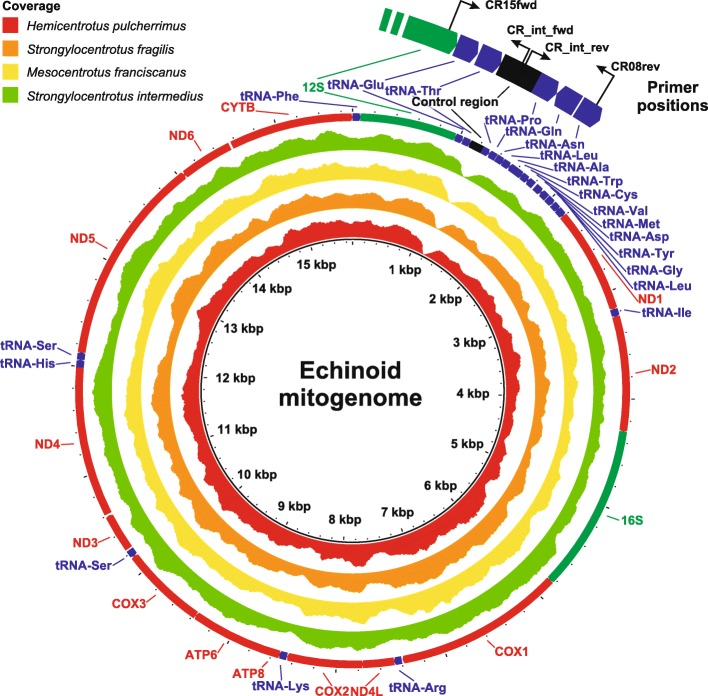
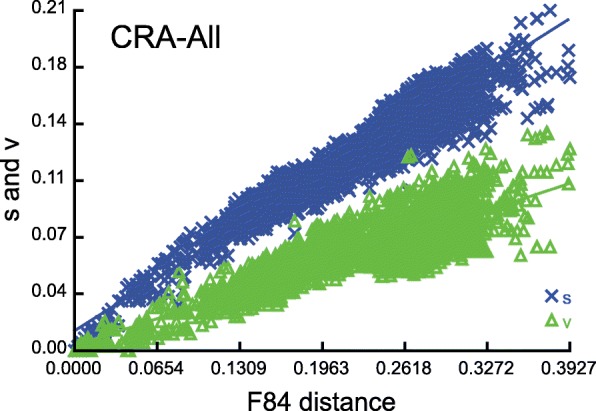
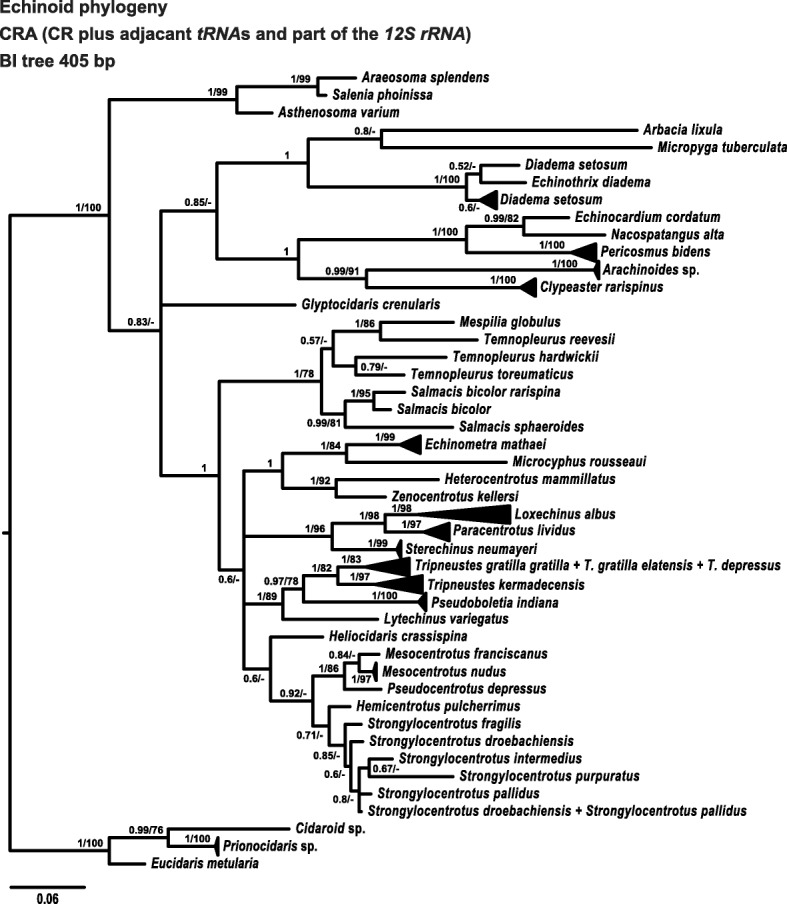
Results: Here we provide novel sequence data for the echinoid mitochondrial control region in over 40 species across the echinoid phylogenetic tree. We demonstrate the advantages of directly targeting the CR and adjacent tRNAs to facilitate complementing low coverage NGS data from complete mitochondrial genome assemblies. Finally, we test the performance of this region as a phylogenetic marker both in the lab and in phylogenetic analyses, and demonstrate its superior performance over the other available mitochondrial markers in echinoids.
Conclusions: Our target region of the mitochondrial CR (1) facilitates the first thorough investigation of this region across a wide range of echinoid taxa, (2) provides a tool for complementing missing data in NGS experiments, and (3) identifies the CR as a powerful, novel marker for phylogenetic inference in echinoids due to its high variability, lack of selection, and high compatibility across the entire class, outperforming conventional mitochondrial markers.
Keywords: Control region; Echinoidea; Mitochondrial markers; Molecular phylogeny; NGS; Sea urchins.
Conflict of interest statement
Ethics approval and consent to participate
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures

Similar articles
-
Characterization of the complete mitochondrial genome of Macrotocinclus affinis (Siluriformes; Loricariidae) and phylogenetic studies of Siluriformes.Mol Biol Rep. 2021 Jan;48(1):677-689. doi: 10.1007/s11033-020-06120-z. Epub 2021 Jan 13. Mol Biol Rep. 2021. PMID: 33442829
-
The influence of molecular markers and methods on inferring the phylogenetic relationships between the representatives of the Arini (parrots, Psittaciformes), determined on the basis of their complete mitochondrial genomes.BMC Evol Biol. 2017 Jul 14;17(1):166. doi: 10.1186/s12862-017-1012-1. BMC Evol Biol. 2017. PMID: 28705202 Free PMC article.
-
Mitochondrial phylogenomics and genetic relationships of closely related pine moth (Lasiocampidae: Dendrolimus) species in China, using whole mitochondrial genomes.BMC Genomics. 2015 Jun 4;16(1):428. doi: 10.1186/s12864-015-1566-5. BMC Genomics. 2015. PMID: 26040695 Free PMC article.
-
Complete mitochondrial genomes of four deep-sea echinoids: conserved mitogenome organization and new insights into the phylogeny and evolution of Echinoidea.PeerJ. 2022 Jul 28;10:e13730. doi: 10.7717/peerj.13730. eCollection 2022. PeerJ. 2022. PMID: 35919401 Free PMC article.
-
A combined morphological and molecular phylogeny for sea urchins (Echinoidea: Echinodermata).Philos Trans R Soc Lond B Biol Sci. 1995 Jan 30;347(1320):213-34. doi: 10.1098/rstb.1995.0023. Philos Trans R Soc Lond B Biol Sci. 1995. PMID: 7746863 Review.
Cited by
-
The first complete mitochondrial genome of the sand dollar Sinaechinocyamus mai (Echinoidea: Clypeasteroida).Genomics. 2020 Mar;112(2):1686-1693. doi: 10.1016/j.ygeno.2019.10.007. Epub 2019 Oct 17. Genomics. 2020. PMID: 31629878 Free PMC article.
-
The Detection and Partial Localisation of Heteroplasmic Mutations in the Mitochondrial Genome of Patients with Diabetic Retinopathy.Int J Mol Sci. 2019 Dec 11;20(24):6259. doi: 10.3390/ijms20246259. Int J Mol Sci. 2019. PMID: 31835862 Free PMC article.
-
First Report of Rickettsia conorii in Hyalomma kumari Ticks.Animals (Basel). 2023 Apr 27;13(9):1488. doi: 10.3390/ani13091488. Animals (Basel). 2023. PMID: 37174525 Free PMC article.
-
A Comparative Analysis of Mitogenomes in Species of the Tapinoma nigerrimum Complex and Other Species of the Genus Tapinoma (Formicidae, Dolichoderinae).Insects. 2024 Dec 2;15(12):957. doi: 10.3390/insects15120957. Insects. 2024. PMID: 39769559 Free PMC article.
-
Distant hybrids of Heliocidaris crassispina (♀) and Strongylocentrotus intermedius (♂): identification and mtDNA heteroplasmy analysis.BMC Evol Biol. 2020 Aug 11;20(1):101. doi: 10.1186/s12862-020-01667-8. BMC Evol Biol. 2020. PMID: 32781979 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
