

Distinct epigenomic patterns are associated with haploinsufficiency and predict risk genes of developmental disorders
- PMID: 29849042
- PMCID: PMC5976622
- DOI: 10.1038/s41467-018-04552-7
Distinct epigenomic patterns are associated with haploinsufficiency and predict risk genes of developmental disorders
Abstract
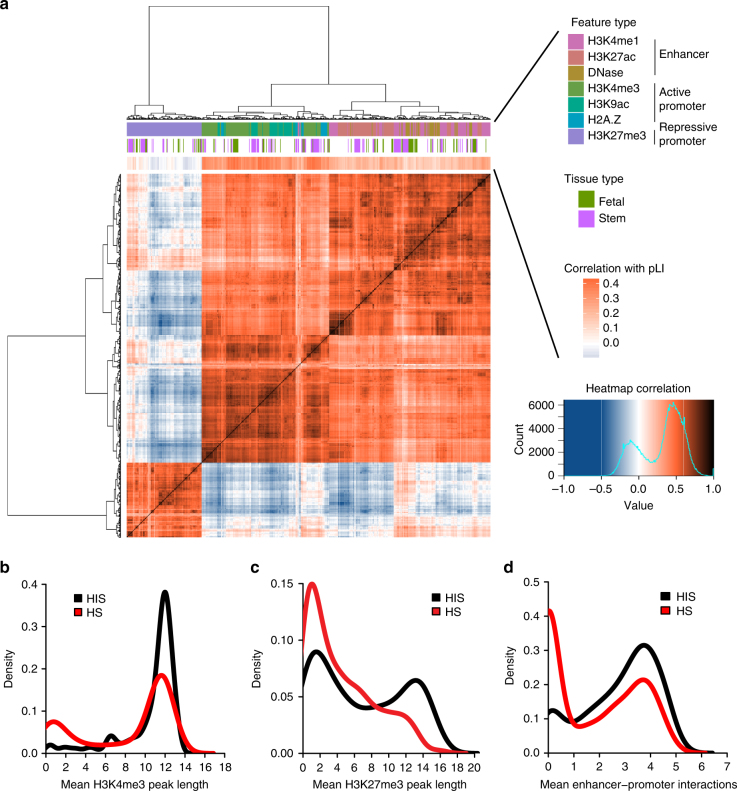
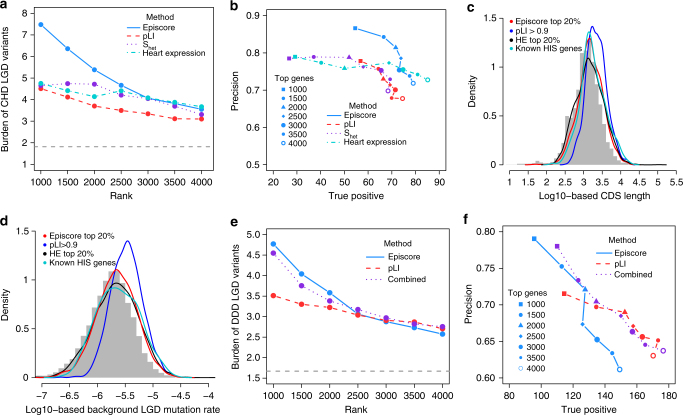
Haploinsufficiency is a major mechanism of genetic risk in developmental disorders. Accurate prediction of haploinsufficient genes is essential for prioritizing and interpreting deleterious variants in genetic studies. Current methods based on mutation intolerance in population data suffer from inadequate power for genes with short transcripts. Here we show haploinsufficiency is strongly associated with epigenomic patterns, and develop a computational method (Episcore) to predict haploinsufficiency leveraging epigenomic data from a broad range of tissue and cell types by machine learning methods. Based on data from recent exome sequencing studies on developmental disorders, Episcore achieves better performance in prioritizing likely-gene-disrupting (LGD) de novo variants than current methods. We further show that Episcore is less-biased by gene size, and complementary to mutation intolerance metrics for prioritizing LGD variants. Our approach enables new applications of epigenomic data and facilitates discovery and interpretation of novel risk variants implicated in developmental disorders.
Conflict of interest statement
The authors declare no competing interests.
Figures


References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
