

Airway Epithelium Dysfunction in Cystic Fibrosis and COPD
- PMID: 29849481
- PMCID: PMC5911336
- DOI: 10.1155/2018/1309746
Airway Epithelium Dysfunction in Cystic Fibrosis and COPD
Abstract
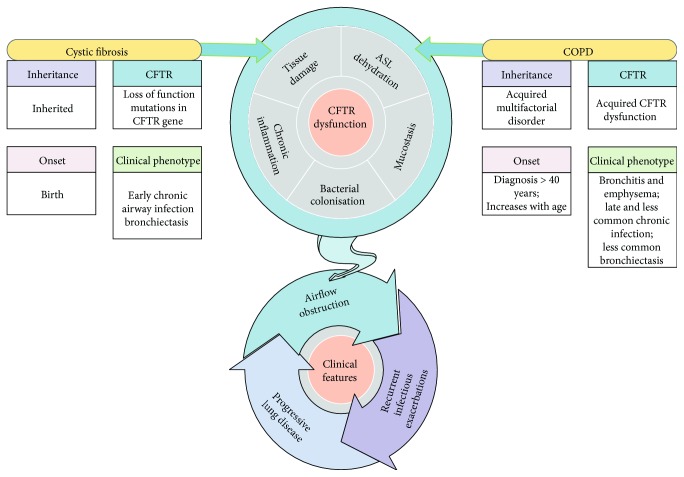
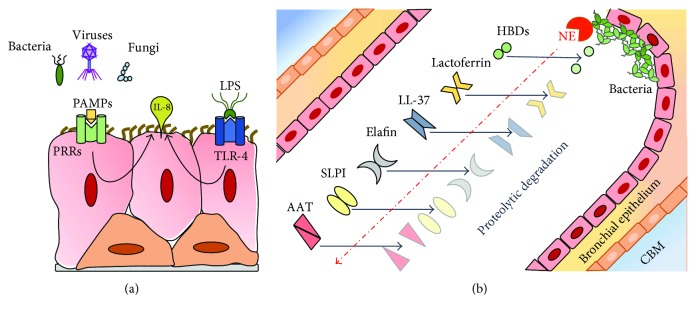
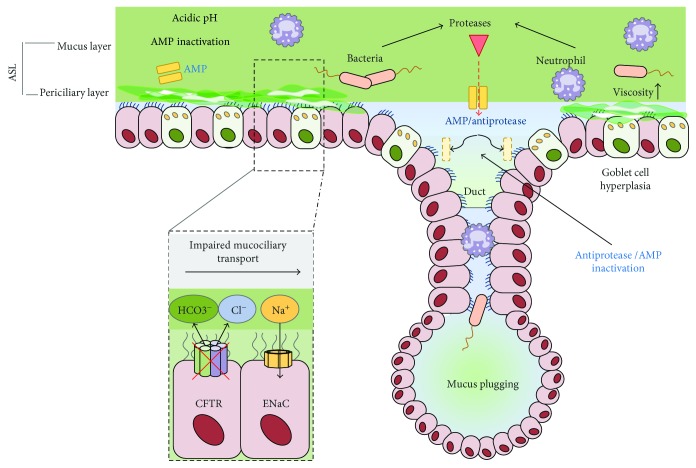
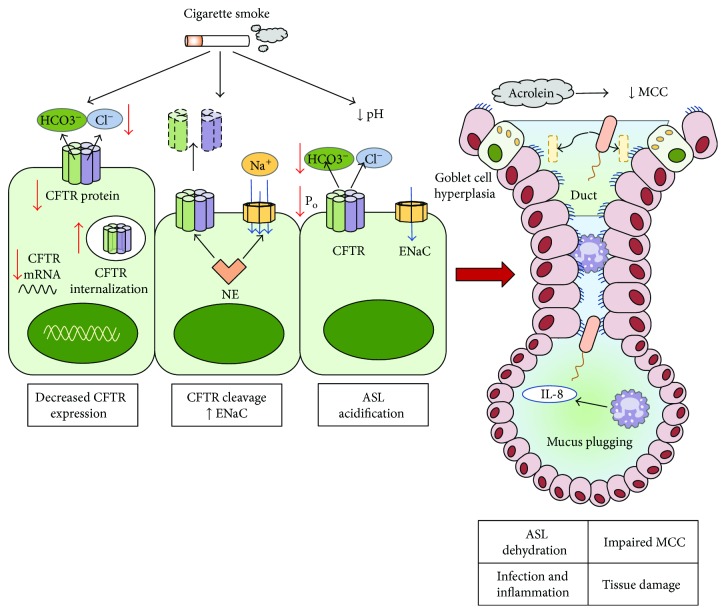
Cystic fibrosis is a genetic disease caused by mutations in the CFTR gene, whereas chronic obstructive pulmonary disease (COPD) is mainly caused by environmental factors (mostly cigarette smoking) on a genetically susceptible background. Although the etiology and pathogenesis of these diseases are different, both are associated with progressive airflow obstruction, airway neutrophilic inflammation, and recurrent exacerbations, suggesting common mechanisms. The airway epithelium plays a crucial role in maintaining normal airway functions. Major molecular and morphologic changes occur in the airway epithelium in both CF and COPD, and growing evidence suggests that airway epithelial dysfunction is involved in disease initiation and progression in both diseases. Structural and functional abnormalities in both airway and alveolar epithelium have a relevant impact on alteration of host defences, immune/inflammatory response, and the repair process leading to progressive lung damage and impaired lung function. In this review, we address the evidence for a critical role of dysfunctional airway epithelial cells in chronic airway inflammation and remodelling in CF and COPD, highlighting the common mechanisms involved in the epithelial dysfunction as well as the similarities and differences of the two diseases.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
