

UMI-count modeling and differential expression analysis for single-cell RNA sequencing
- PMID: 29855333
- PMCID: PMC5984373
- DOI: 10.1186/s13059-018-1438-9
UMI-count modeling and differential expression analysis for single-cell RNA sequencing
Abstract
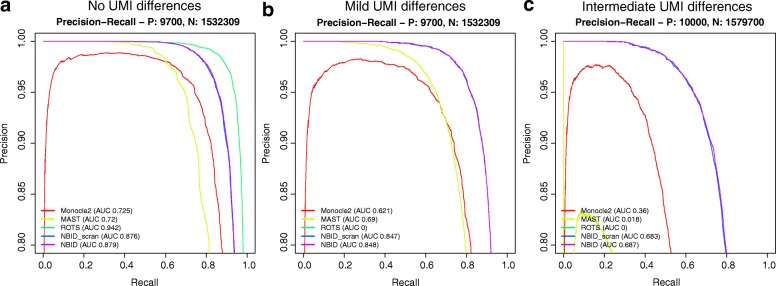
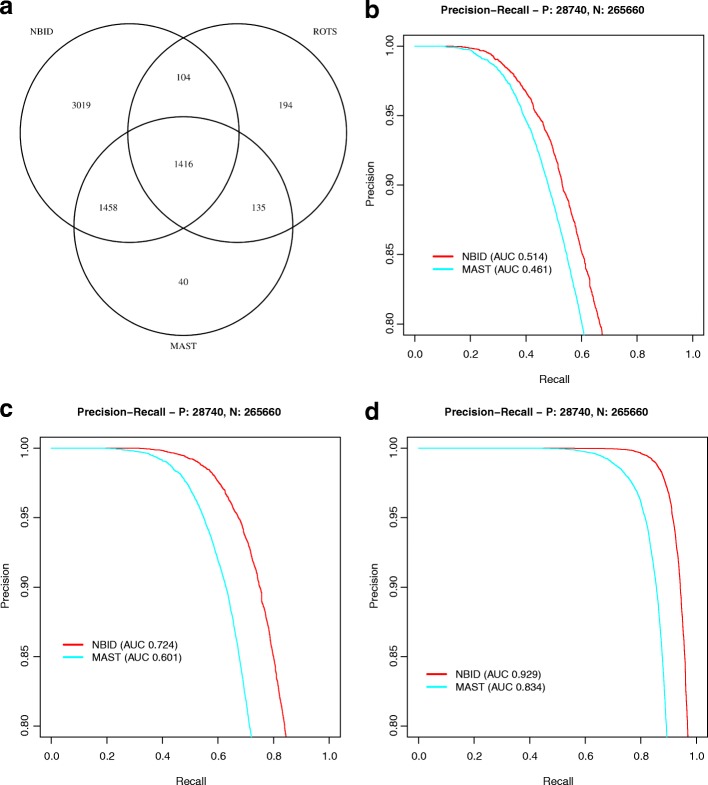
Read counting and unique molecular identifier (UMI) counting are the principal gene expression quantification schemes used in single-cell RNA-sequencing (scRNA-seq) analysis. By using multiple scRNA-seq datasets, we reveal distinct distribution differences between these schemes and conclude that the negative binomial model is a good approximation for UMI counts, even in heterogeneous populations. We further propose a novel differential expression analysis algorithm based on a negative binomial model with independent dispersions in each group (NBID). Our results show that this properly controls the FDR and achieves better power for UMI counts when compared to other recently developed packages for scRNA-seq analysis.
Keywords: Differential expression analysis; Negative binomial; Unique molecular identifier.
Conflict of interest statement
Ethics approval and consent to participate
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
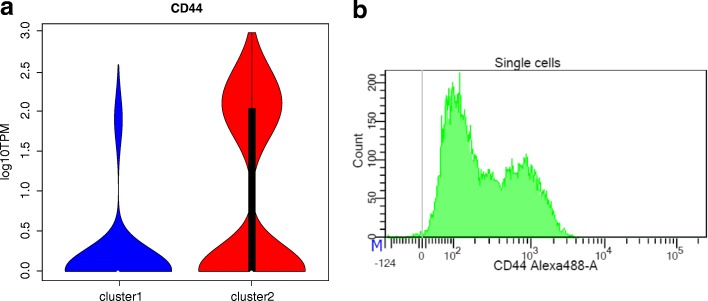
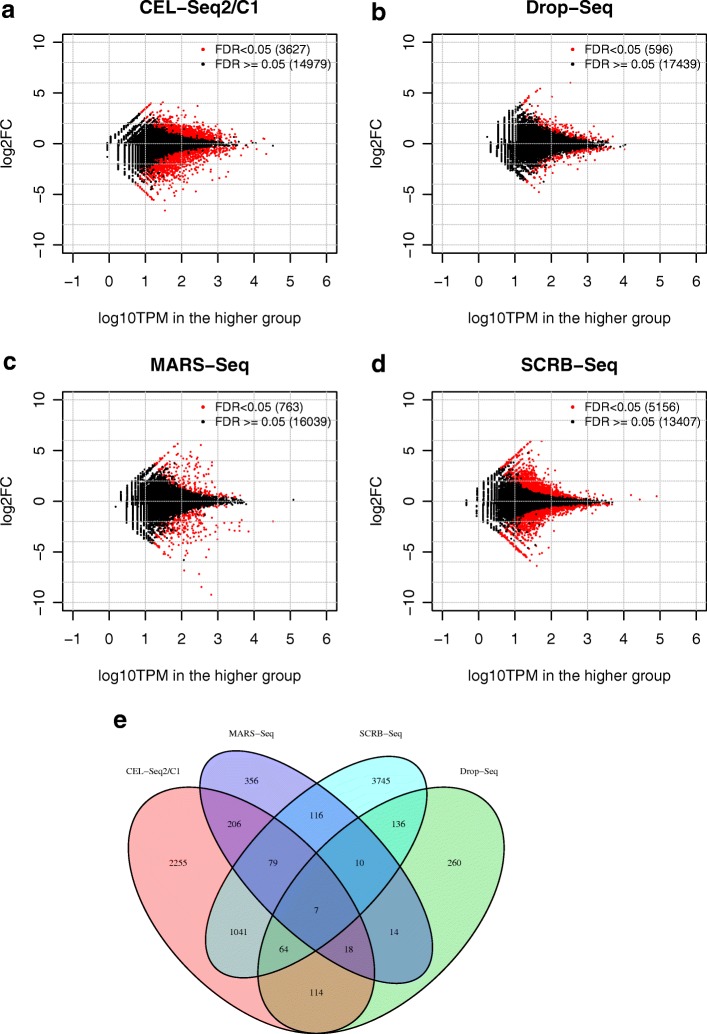
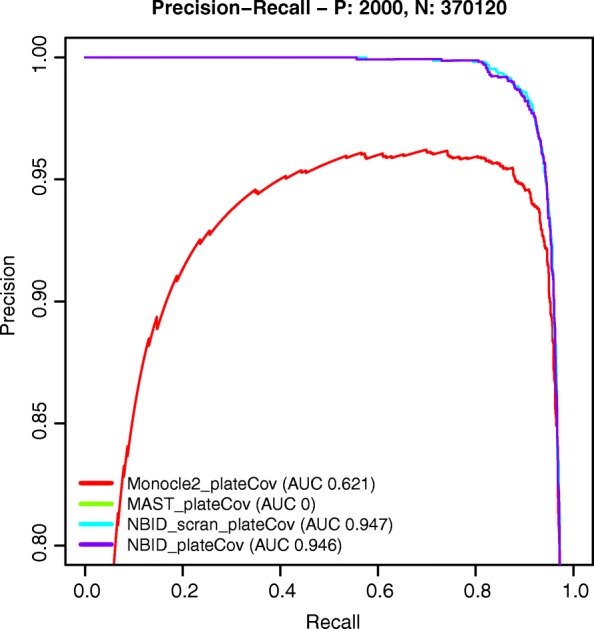
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
