

Genetic Inactivation of CD33 in Hematopoietic Stem Cells to Enable CAR T Cell Immunotherapy for Acute Myeloid Leukemia
- PMID: 29856956
- PMCID: PMC6003425
- DOI: 10.1016/j.cell.2018.05.013
Genetic Inactivation of CD33 in Hematopoietic Stem Cells to Enable CAR T Cell Immunotherapy for Acute Myeloid Leukemia
Abstract
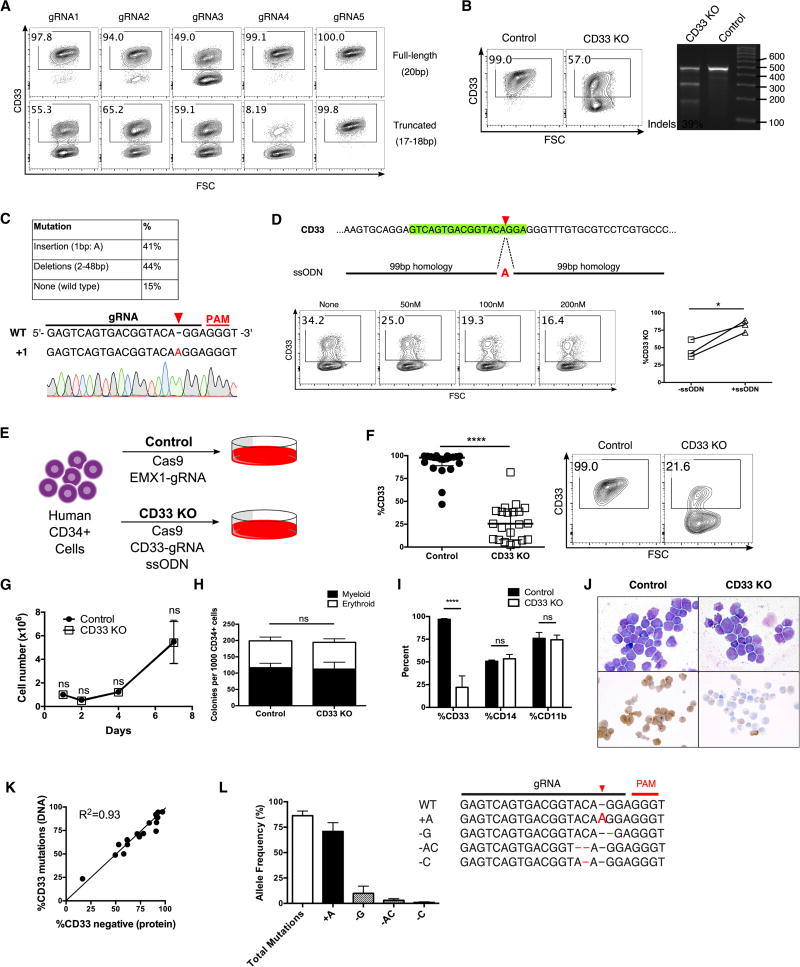
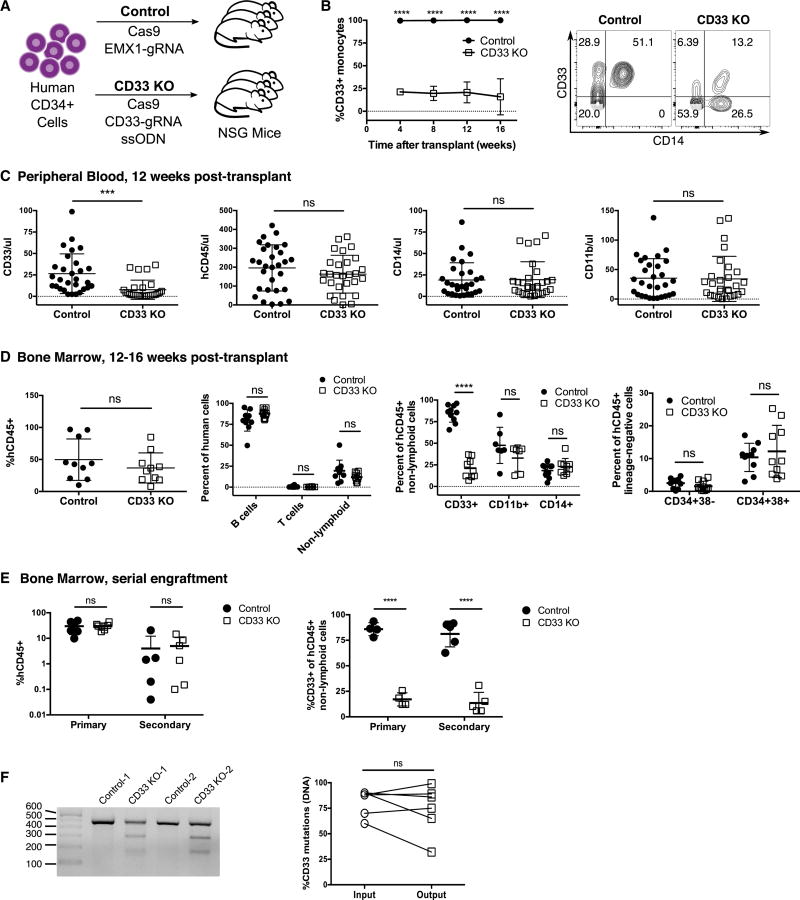
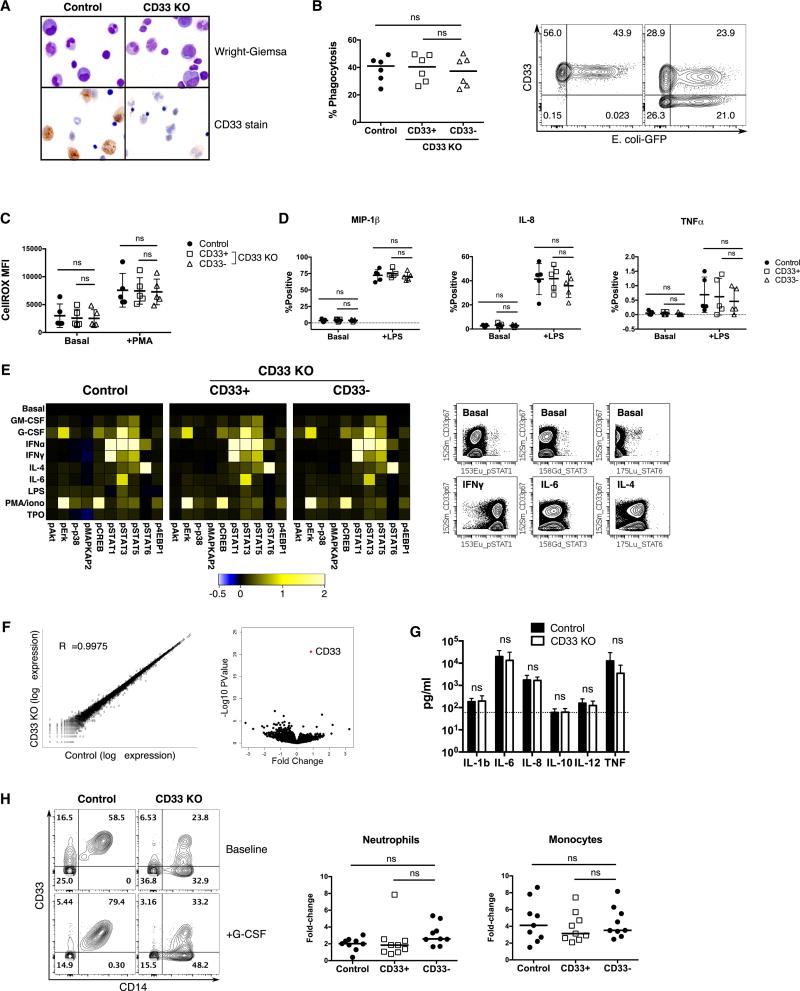
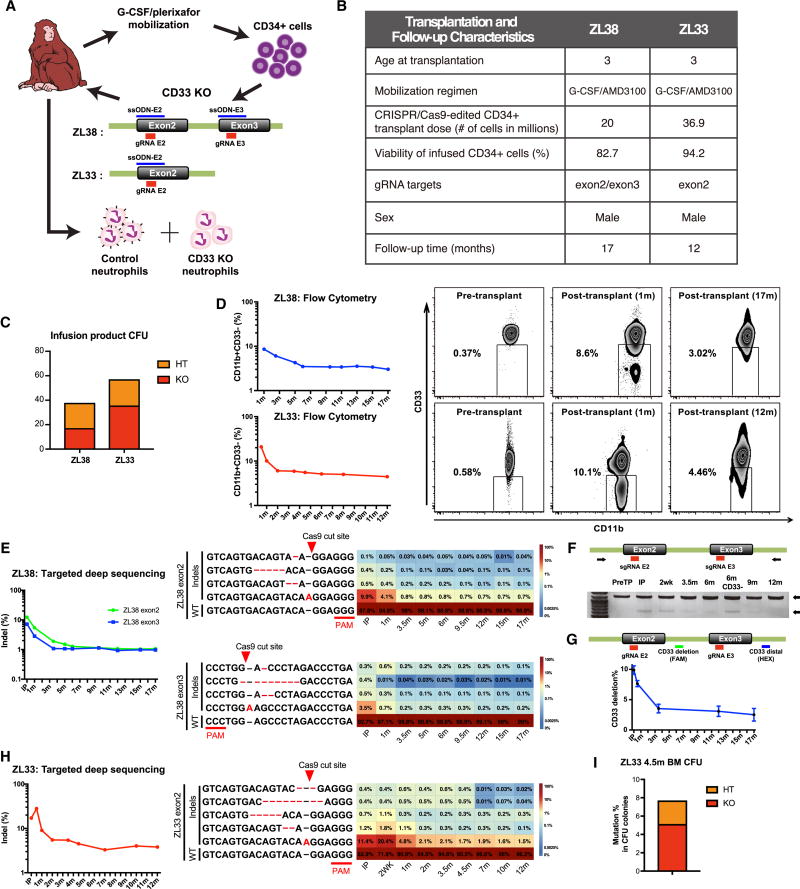
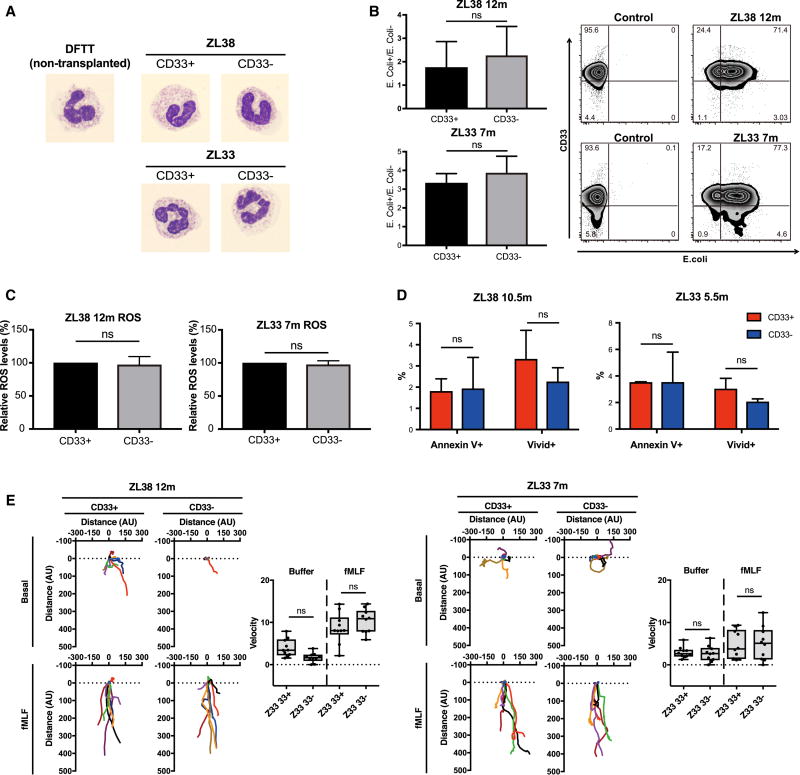
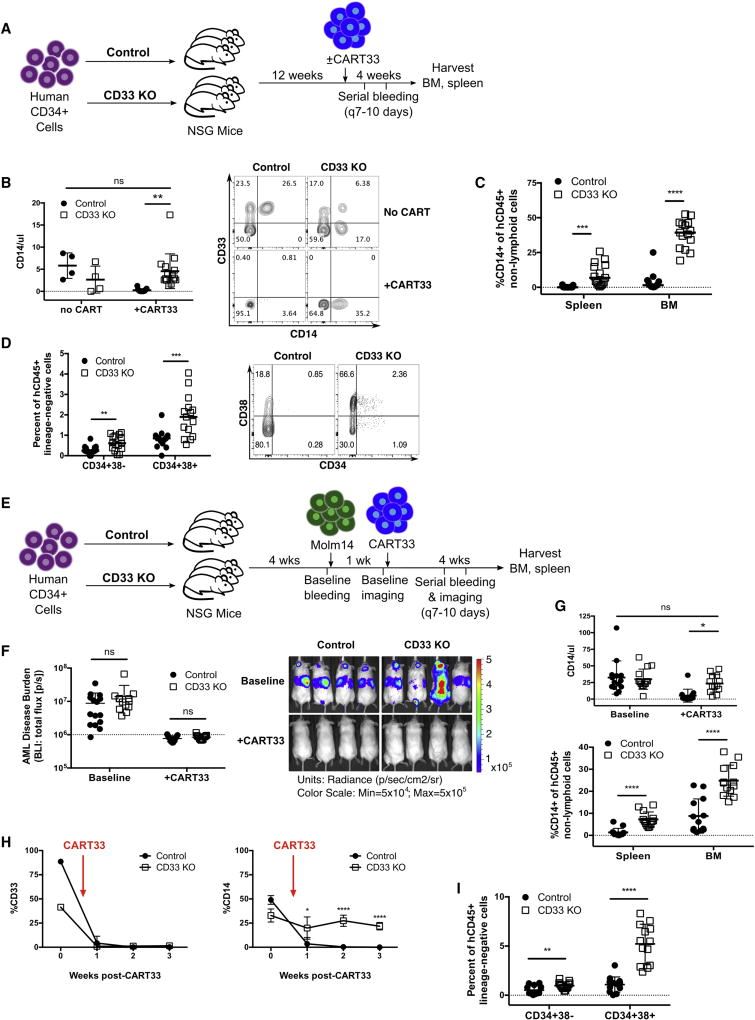
The absence of cancer-restricted surface markers is a major impediment to antigen-specific immunotherapy using chimeric antigen receptor (CAR) T cells. For example, targeting the canonical myeloid marker CD33 in acute myeloid leukemia (AML) results in toxicity from destruction of normal myeloid cells. We hypothesized that a leukemia-specific antigen could be created by deleting CD33 from normal hematopoietic stem and progenitor cells (HSPCs), thereby generating a hematopoietic system resistant to CD33-targeted therapy and enabling specific targeting of AML with CAR T cells. We generated CD33-deficient human HSPCs and demonstrated normal engraftment and differentiation in immunodeficient mice. Autologous CD33 KO HSPC transplantation in rhesus macaques demonstrated long-term multilineage engraftment of gene-edited cells with normal myeloid function. CD33-deficient cells were impervious to CD33-targeting CAR T cells, allowing for efficient elimination of leukemia without myelotoxicity. These studies illuminate a novel approach to antigen-specific immunotherapy by genetically engineering the host to avoid on-target, off-tumor toxicity.
Keywords: CD33; CRISPR/Cas9 gene editing; acute myeloid leukemia; chimeric antigen receptor T cells; hematopoiesis; immunotherapy; non-human primate hematopoiesis.
Copyright © 2018 Elsevier Inc. All rights reserved.
Figures
References
-
- Appelbaum FR, Bernstein ID. Gemtuzumab ozogamicin for acute myeloid leukemia. Blood. 2017;130:2373. - PubMed
-
- Biasco L, Pellin D, Scala S, Dionisio F, Basso-Ricci L, Leonardelli L, Scaramuzza S, Baricordi C, Ferrua F, Cicalese MP, et al. In Vivo Tracking of Human Hematopoiesis Reveals Patterns of Clonal Dynamics during Early and Steady-State Reconstitution Phases. Cell Stem Cell. 2016;19:107–119. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
