

The Role of JMY in p53 Regulation
- PMID: 29857553
- PMCID: PMC6025294
- DOI: 10.3390/cancers10060173
The Role of JMY in p53 Regulation
Abstract
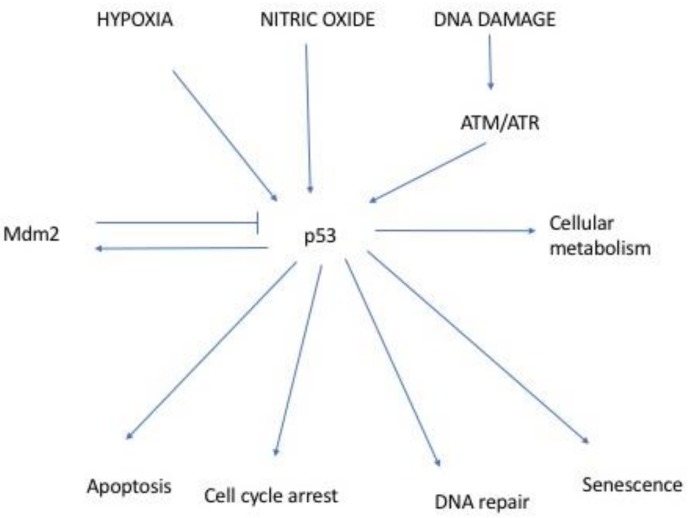
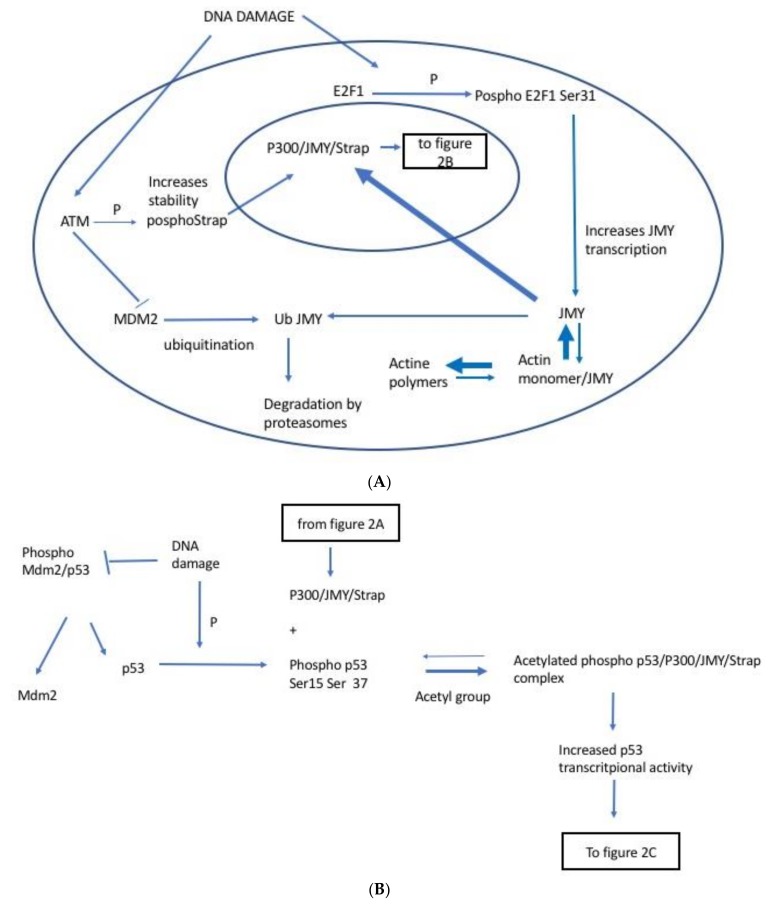
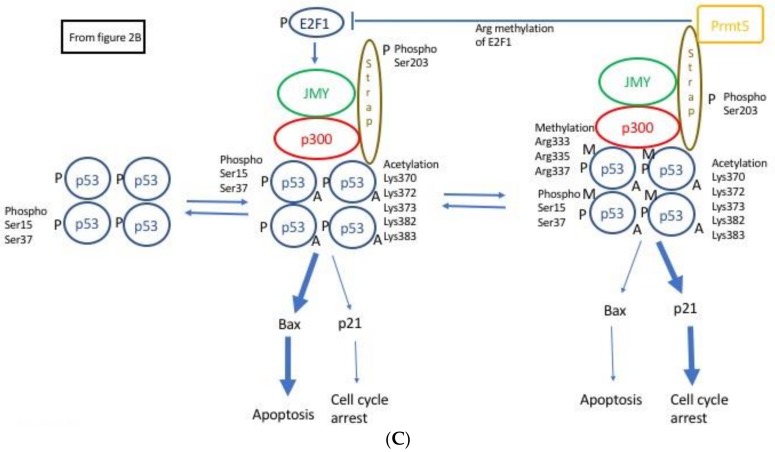
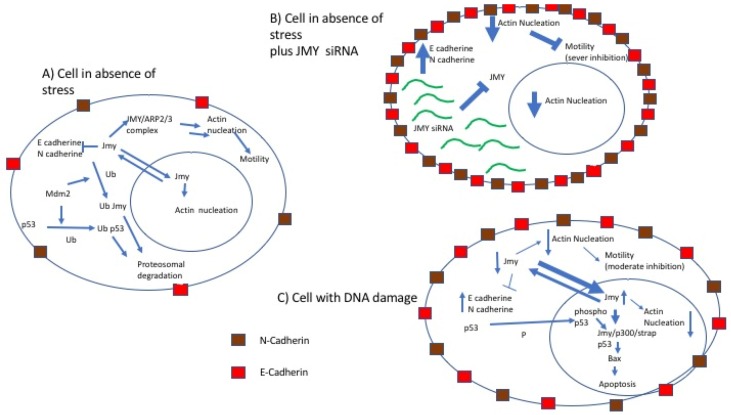
Following the event of DNA damage, the level of tumour suppressor protein p53 increases inducing either cell cycle arrest or apoptosis. Junctional Mediating and Regulating Y protein (JMY) is a transcription co-factor involved in p53 regulation. In event of DNA damage, JMY levels also upregulate in the nucleus where JMY forms a co-activator complex with p300/CREB-binding protein (p300/CBP), Apoptosis-stimulating protein of p53 (ASPP) and Stress responsive activator of p53 (Strap). This co-activator complex then binds to and increases the ability of p53 to induce transcription of proteins triggering apoptosis but not cell cycle arrest. This then suggests that the increase of JMY levels due to DNA damage putatively "directs" p53 activity toward triggering apoptosis. JMY expression is also linked to increased cell motility as it: (1) downregulates the expression of adhesion molecules of the Cadherin family and (2) induces actin nucleation, making cells less adhesive and more mobile, favouring metastasis. All these characteristics taken together imply that JMY possesses both tumour suppressive and tumour metastasis promoting capabilities.
Keywords: JMY; apoptosis; motility; p53; regulation.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
