

Vex-seq: high-throughput identification of the impact of genetic variation on pre-mRNA splicing efficiency
- PMID: 29859120
- PMCID: PMC5984807
- DOI: 10.1186/s13059-018-1437-x
Vex-seq: high-throughput identification of the impact of genetic variation on pre-mRNA splicing efficiency
Abstract
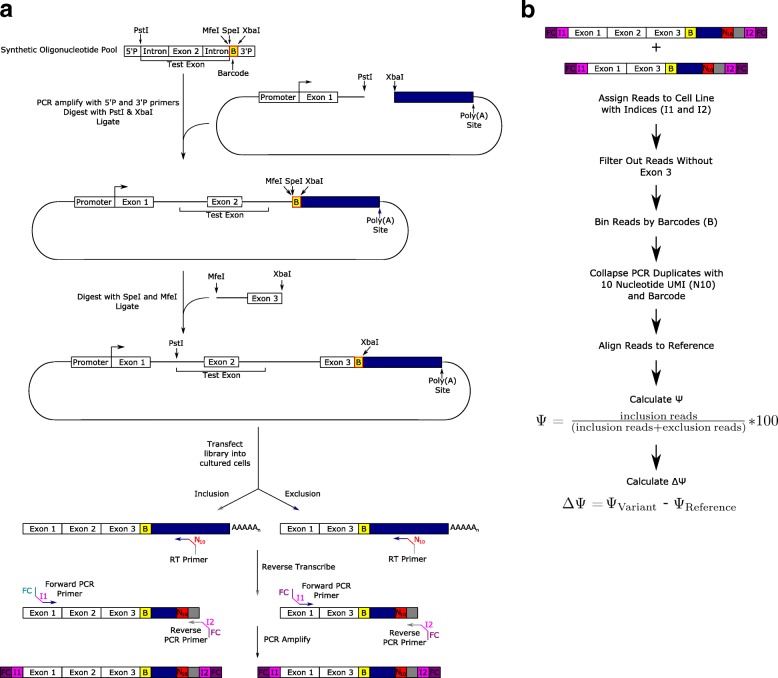
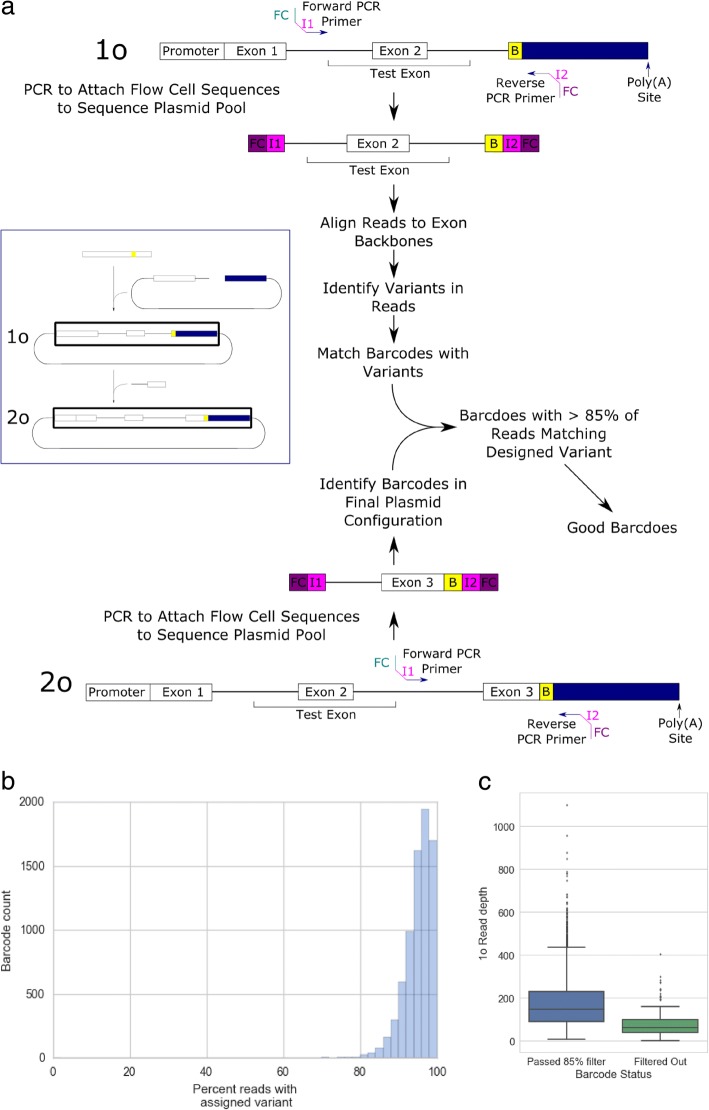
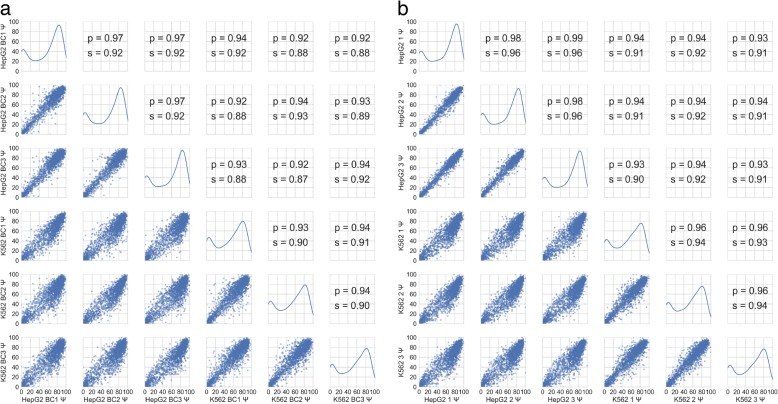
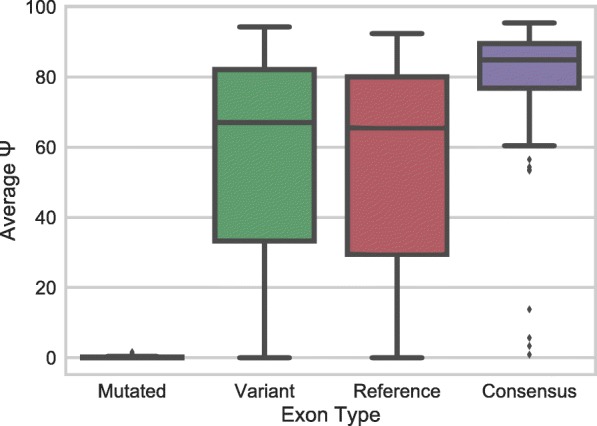
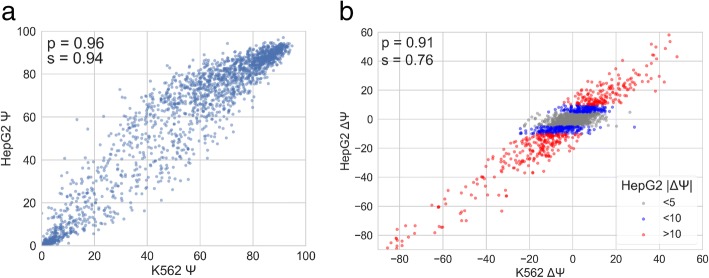
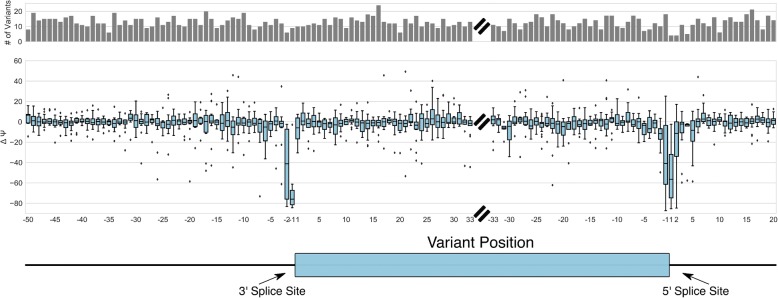
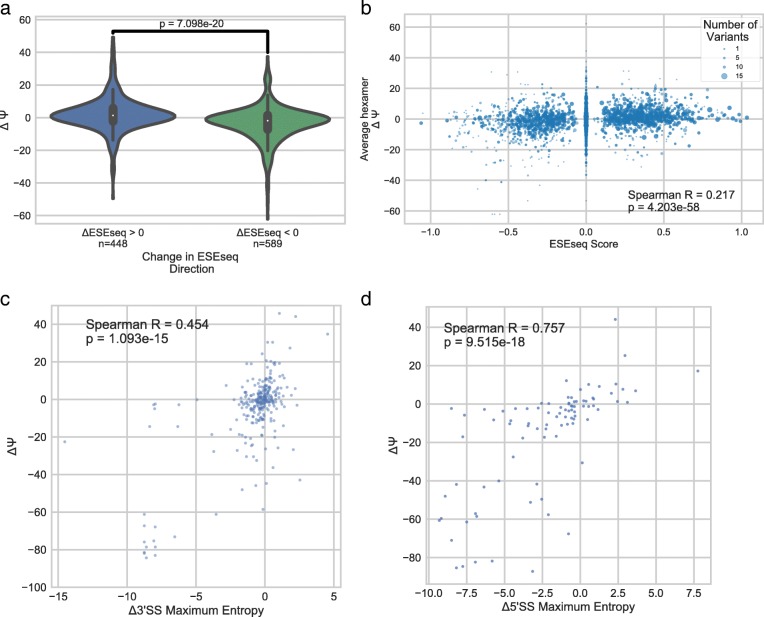
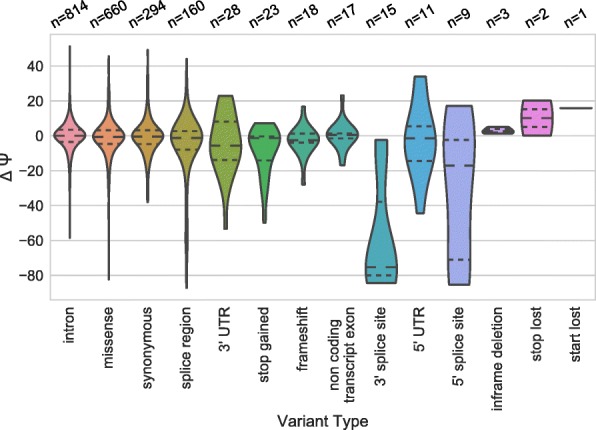
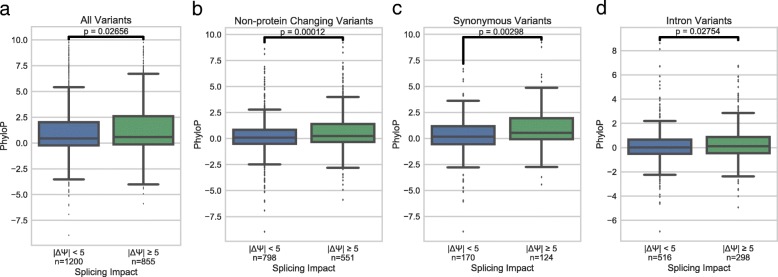
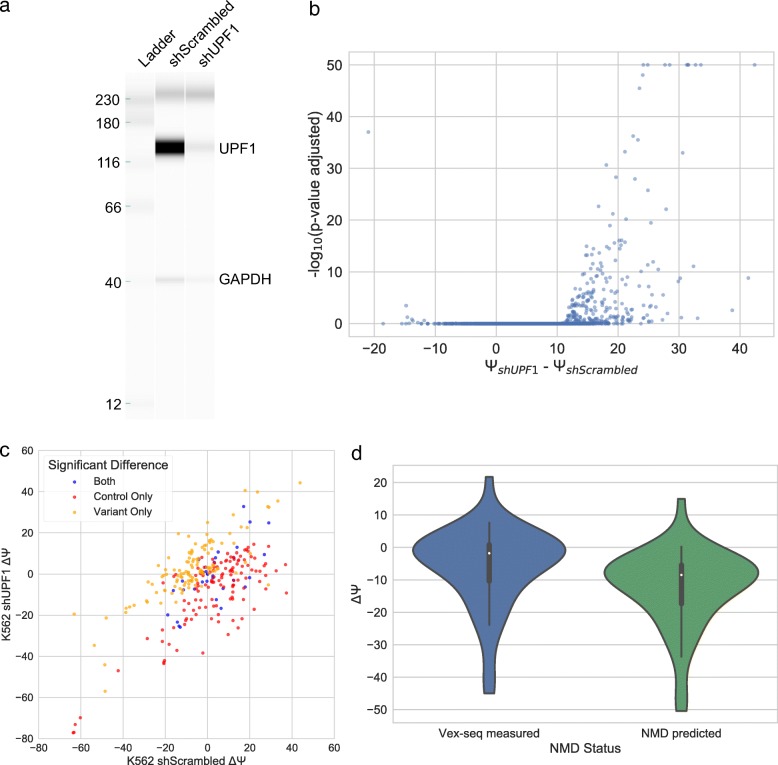
Understanding the functional impact of genomic variants is a major goal of modern genetics and personalized medicine. Although many synonymous and non-coding variants act through altering the efficiency of pre-mRNA splicing, it is difficult to predict how these variants impact pre-mRNA splicing. Here, we describe a massively parallel approach we use to test the impact on pre-mRNA splicing of 2059 human genetic variants spanning 110 alternative exons. This method, called variant exon sequencing (Vex-seq), yields data that reinforce known mechanisms of pre-mRNA splicing, identifies variants that impact pre-mRNA splicing, and will be useful for increasing our understanding of genome function.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
