

De Novo and Inherited Loss-of-Function Variants in TLK2: Clinical and Genotype-Phenotype Evaluation of a Distinct Neurodevelopmental Disorder
- PMID: 29861108
- PMCID: PMC5992133
- DOI: 10.1016/j.ajhg.2018.04.014
De Novo and Inherited Loss-of-Function Variants in TLK2: Clinical and Genotype-Phenotype Evaluation of a Distinct Neurodevelopmental Disorder
Abstract
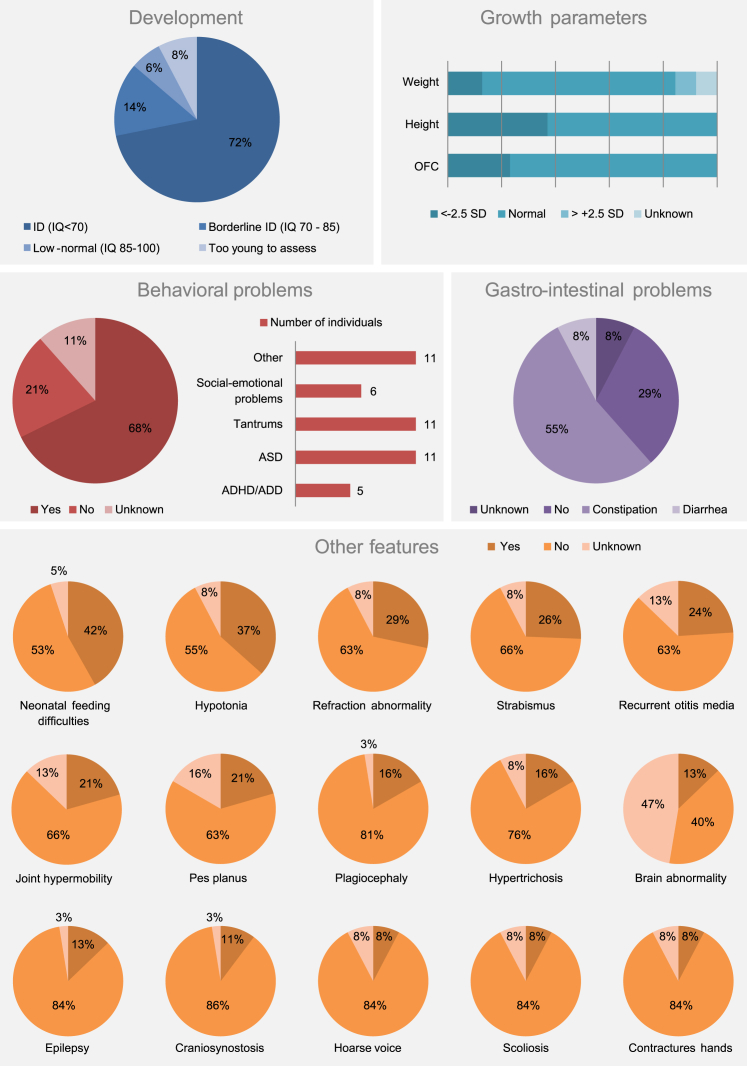
Next-generation sequencing is a powerful tool for the discovery of genes related to neurodevelopmental disorders (NDDs). Here, we report the identification of a distinct syndrome due to de novo or inherited heterozygous mutations in Tousled-like kinase 2 (TLK2) in 38 unrelated individuals and two affected mothers, using whole-exome and whole-genome sequencing technologies, matchmaker databases, and international collaborations. Affected individuals had a consistent phenotype, characterized by mild-borderline neurodevelopmental delay (86%), behavioral disorders (68%), severe gastro-intestinal problems (63%), and facial dysmorphism including blepharophimosis (82%), telecanthus (74%), prominent nasal bridge (68%), broad nasal tip (66%), thin vermilion of the upper lip (62%), and upslanting palpebral fissures (55%). Analysis of cell lines from three affected individuals showed that mutations act through a loss-of-function mechanism in at least two case subjects. Genotype-phenotype analysis and comparison of computationally modeled faces showed that phenotypes of these and other individuals with loss-of-function variants significantly overlapped with phenotypes of individuals with other variant types (missense and C-terminal truncating). This suggests that haploinsufficiency of TLK2 is the most likely underlying disease mechanism, leading to a consistent neurodevelopmental phenotype. This work illustrates the power of international data sharing, by the identification of 40 individuals from 26 different centers in 7 different countries, allowing the identification, clinical delineation, and genotype-phenotype evaluation of a distinct NDD caused by mutations in TLK2.
Keywords: Tousled-like; facial averaging; haploinsufficiency; intellectual disability; kinase.
Copyright © 2018 The Authors. Published by Elsevier Inc. All rights reserved.
Figures



References
-
- Lelieveld S.H., Reijnders M.R., Pfundt R., Yntema H.G., Kamsteeg E.J., de Vries P., de Vries B.B., Willemsen M.H., Kleefstra T., Löhner K. Meta-analysis of 2,104 trios provides support for 10 new genes for intellectual disability. Nat. Neurosci. 2016;19:1194–1196. - PubMed
-
- American Psychiatric Association (2013). Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (Washington, DC).
-
- Yamakawa A., Kameoka Y., Hashimoto K., Yoshitake Y., Nishikawa K., Tanihara K., Date T. cDNA cloning and chromosomal mapping of genes encoding novel protein kinases termed PKU-alpha and PKU-beta, which have nuclear localization signal. Gene. 1997;202:193–201. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
