

Tumor Necrosis Factor and Interleukin-1 β Modulate Synaptic Plasticity during Neuroinflammation
- PMID: 29861718
- PMCID: PMC5976900
- DOI: 10.1155/2018/8430123
Tumor Necrosis Factor and Interleukin-1 β Modulate Synaptic Plasticity during Neuroinflammation
Abstract
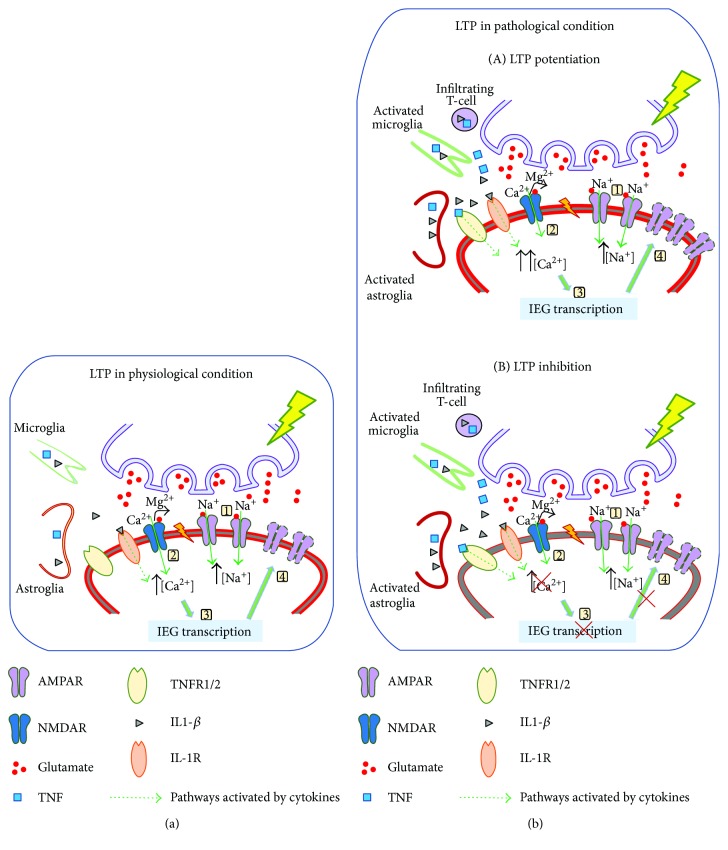
Cytokines are constitutively released in the healthy brain by resident myeloid cells to keep proper synaptic plasticity, either in the form of Hebbian synaptic plasticity or of homeostatic plasticity. However, when cytokines dramatically increase, establishing a status of neuroinflammation, the synaptic action of such molecules remarkably interferes with brain circuits of learning and cognition and contributes to excitotoxicity and neurodegeneration. Among others, interleukin-1β (IL-1β) and tumor necrosis factor (TNF) are the best studied proinflammatory cytokines in both physiological and pathological conditions and have been invariably associated with long-term potentiation (LTP) (Hebbian synaptic plasticity) and synaptic scaling (homeostatic plasticity), respectively. Multiple sclerosis (MS) is the prototypical neuroinflammatory disease, in which inflammation triggers excitotoxic mechanisms contributing to neurodegeneration. IL-β and TNF are increased in the brain of MS patients and contribute to induce the changes in synaptic plasticity occurring in MS patients and its animal model, the experimental autoimmune encephalomyelitis (EAE). This review will introduce and discuss current evidence of the role of IL-1β and TNF in the regulation of synaptic strength at both physiological and pathological levels, in particular speculating on their involvement in the synaptic plasticity changes observed in the EAE brain.
Figures

References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
