

Destabilizing NEK2 overcomes resistance to proteasome inhibition in multiple myeloma
- PMID: 29863498
- PMCID: PMC6026005
- DOI: 10.1172/JCI98765
Destabilizing NEK2 overcomes resistance to proteasome inhibition in multiple myeloma
Abstract
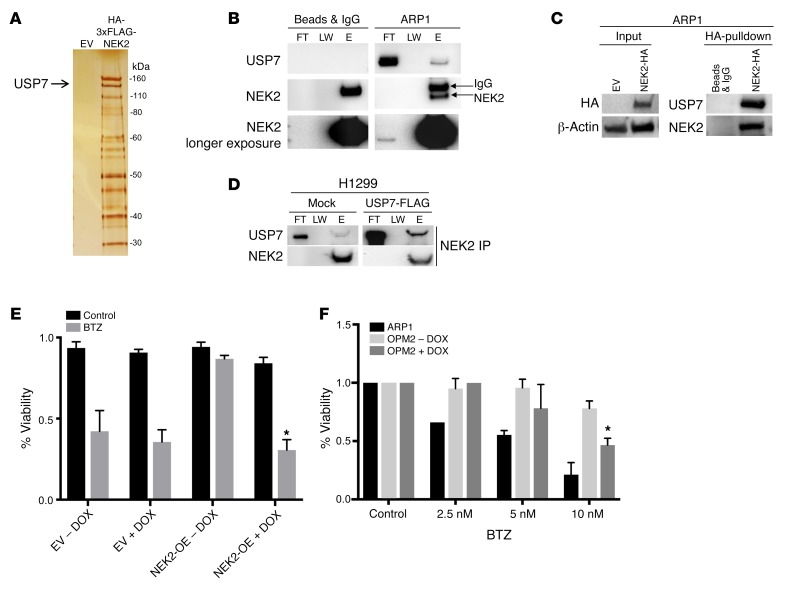
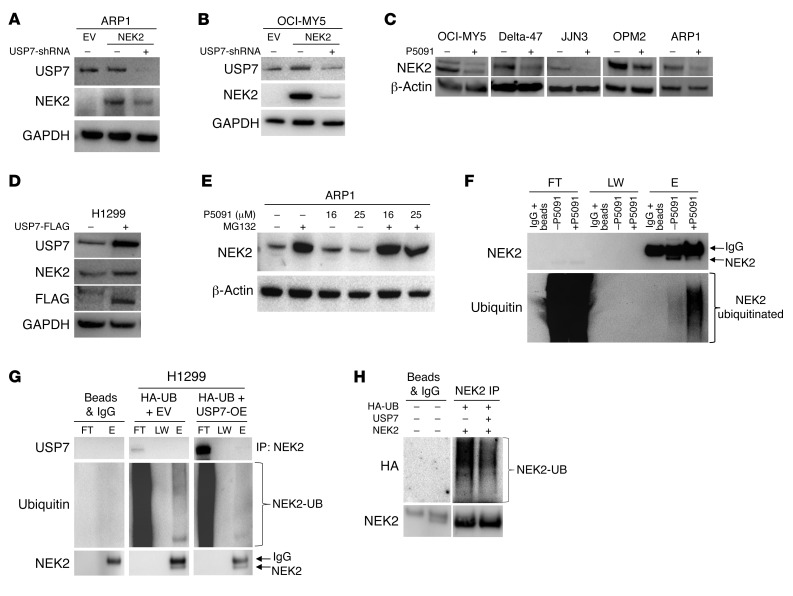
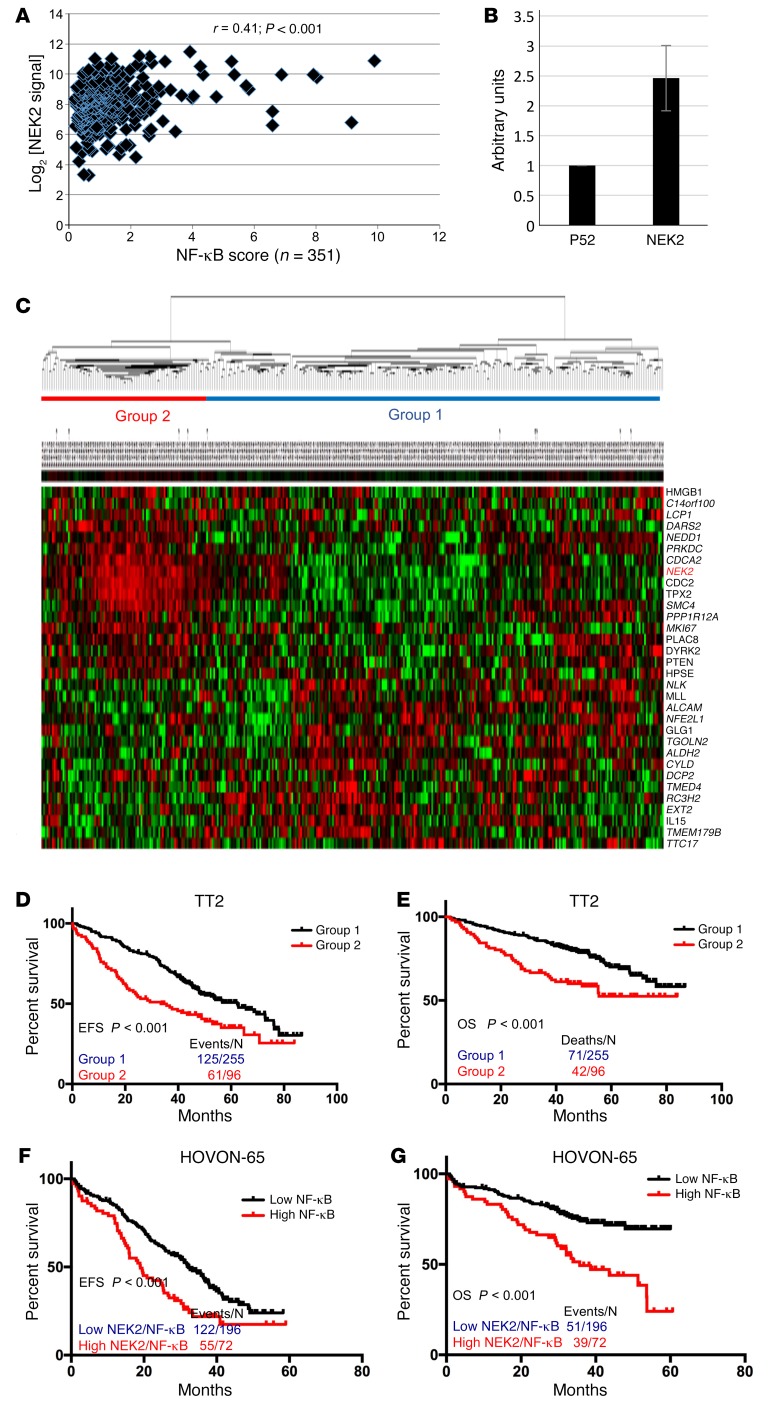
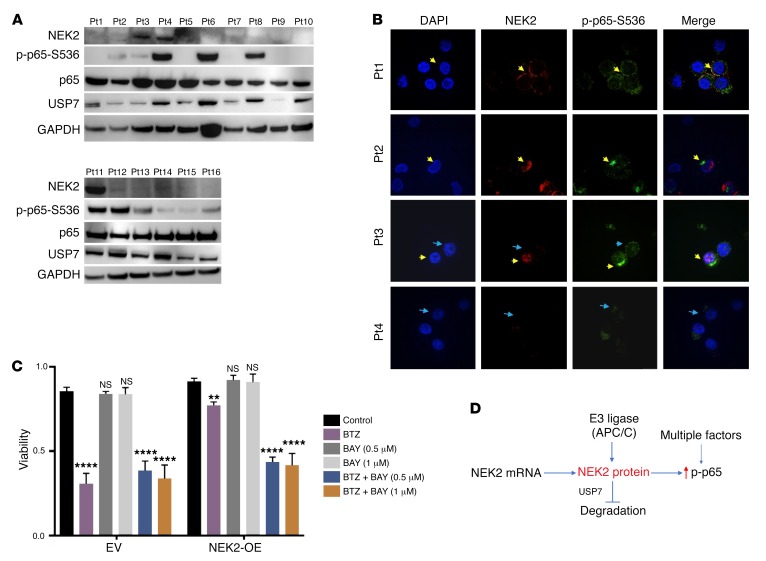
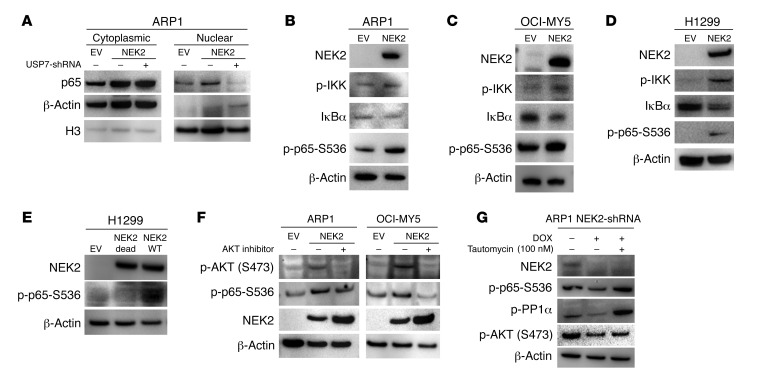
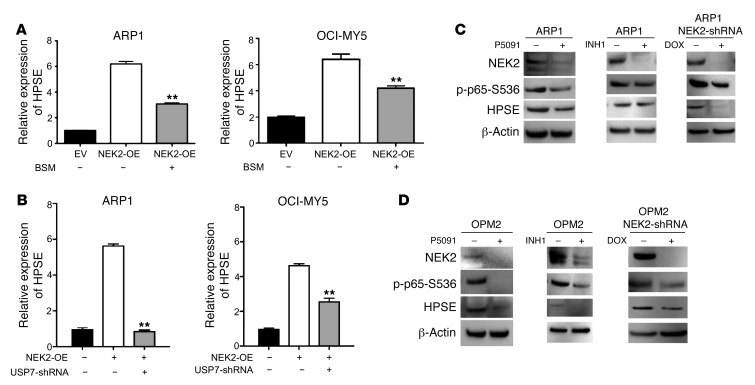
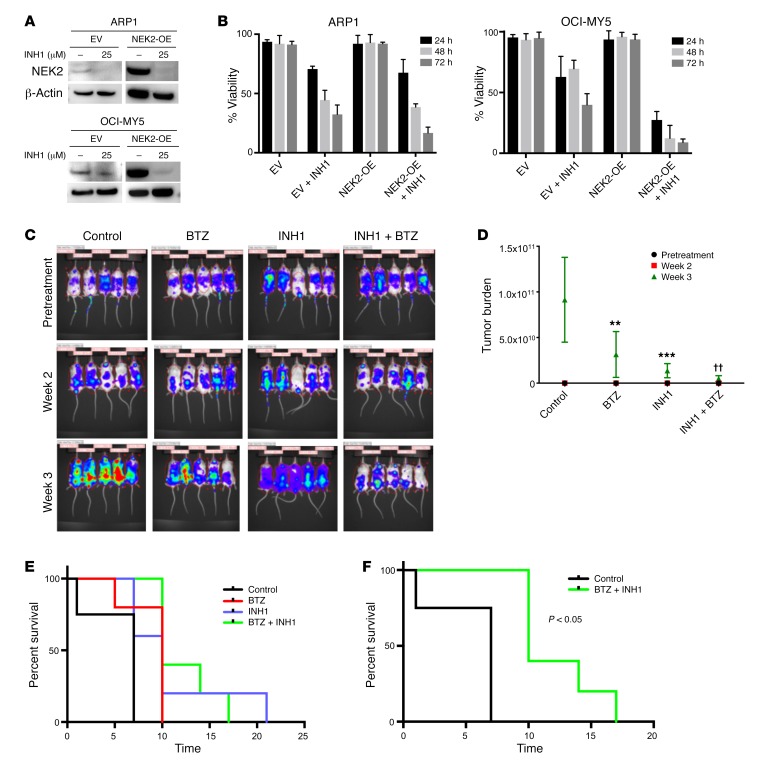
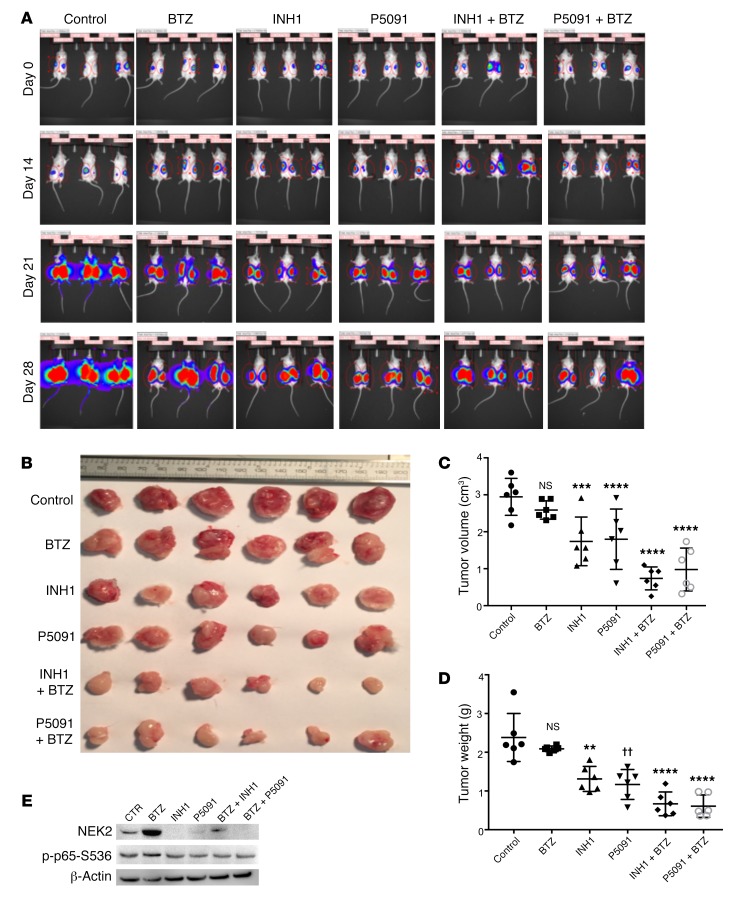
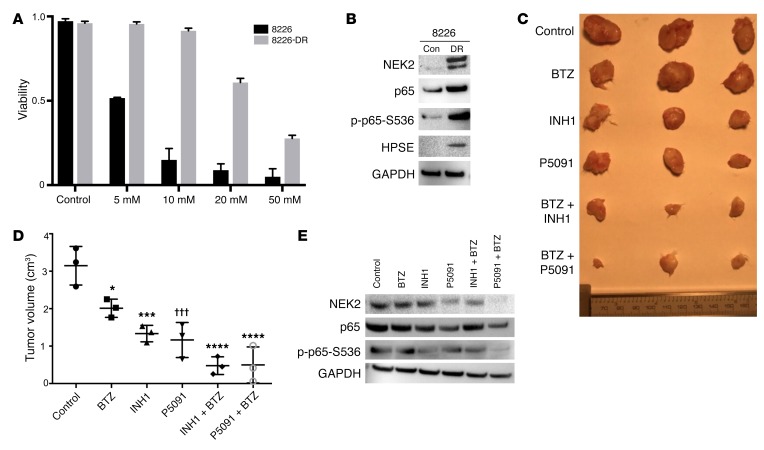
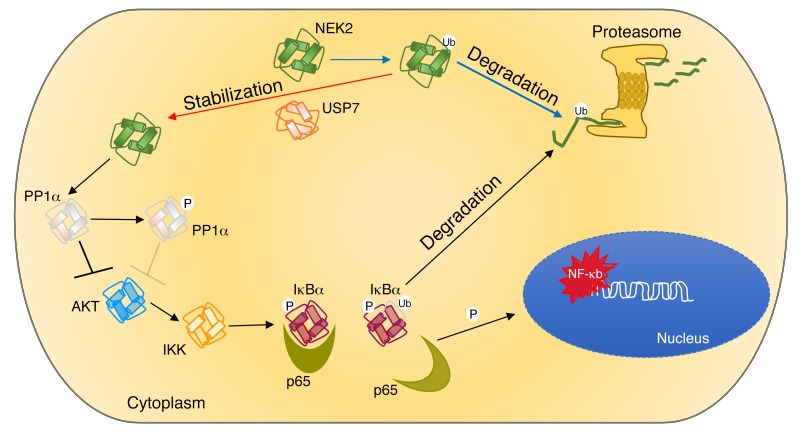
Drug resistance remains the key problem in cancer treatment. It is now accepted that each myeloma patient harbors multiple subclones and subclone dominance may change over time. The coexistence of multiple subclones with high or low chromosomal instability (CIN) signature causes heterogeneity and drug resistance with consequent disease relapse. In this study, using a tandem affinity purification-mass spectrometry (TAP-MS) technique, we found that NEK2, a CIN gene, was bound to the deubiquitinase USP7. Binding to USP7 prevented NEK2 ubiquitination resulting in NEK2 stabilization. Increased NEK2 kinase levels activated the canonical NF-κB signaling pathway through the PP1α/AKT axis. Newly diagnosed myeloma patients with activated NF-κB signaling through increased NEK2 activity had poorer event-free and overall survivals based on multiple independent clinical cohorts. We also found that NEK2 activated heparanase, a secreted enzyme, responsible for bone destruction in an NF-κB-dependent manner. Intriguingly, both NEK2 and USP7 inhibitors showed great efficacy in inhibiting myeloma cell growth and overcoming NEK2-induced and -acquired drug resistance in xenograft myeloma mouse models.
Keywords: B cells; Cancer; Oncology.
Conflict of interest statement
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
