

Linking hnRNP Function to ALS and FTD Pathology
- PMID: 29867335
- PMCID: PMC5962818
- DOI: 10.3389/fnins.2018.00326
Linking hnRNP Function to ALS and FTD Pathology
Abstract
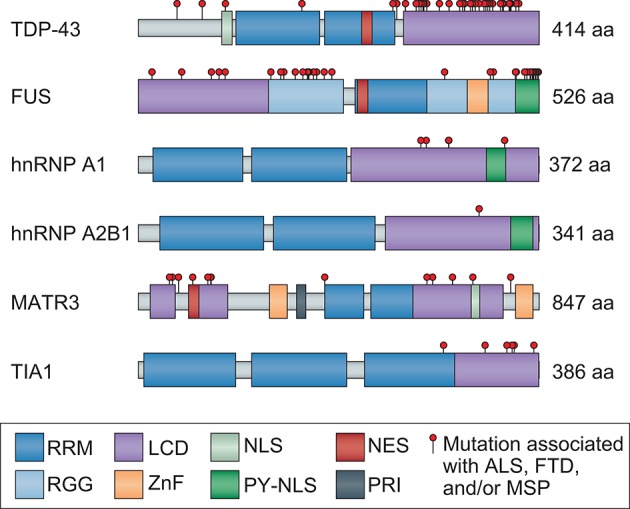
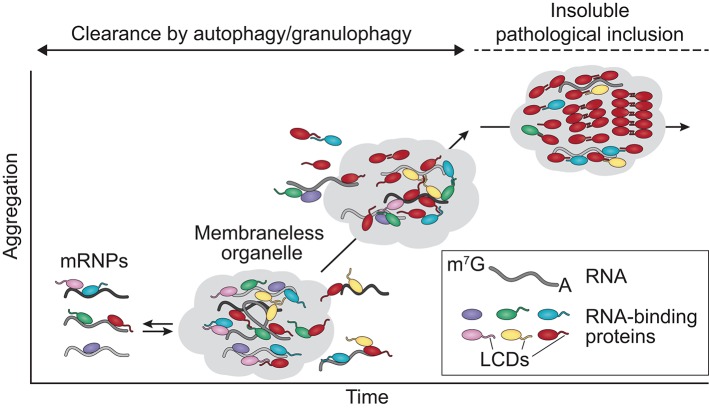
Following years of rapid progress identifying the genetic underpinnings of amyotrophic lateral sclerosis (ALS) and related diseases such as frontotemporal dementia (FTD), remarkable consistencies have emerged pointing to perturbed biology of heterogeneous nuclear ribonucleoproteins (hnRNPs) as a central driver of pathobiology. To varying extents these RNA-binding proteins are deposited in pathological inclusions in affected tissues in ALS and FTD. Moreover, mutations in hnRNPs account for a significant number of familial cases of ALS and FTD. Here we review the normal function and potential pathogenic contribution of TDP-43, FUS, hnRNP A1, hnRNP A2B1, MATR3, and TIA1 to disease. We highlight recent evidence linking the low complexity sequence domains (LCDs) of these hnRNPs to the formation of membraneless organelles and discuss how alterations in the dynamics of these organelles could contribute to disease. In particular, we discuss the various roles of disease-associated hnRNPs in stress granule assembly and disassembly, and examine the emerging hypothesis that disease-causing mutations in these proteins lead to accumulation of persistent stress granules.
Keywords: amyotrophic lateral sclerosis; frontotemporal dementia; hnRNPs; membraneless organelles; stress granules.
Figures
References
-
- Austin J. A., Wright G. S., Watanabe S., Grossmann J. G., Antonyuk S. V., Yamanaka K., et al. . (2014). Disease causing mutants of TDP-43 nucleic acid binding domains are resistant to aggregation and have increased stability and half-life. Proc. Natl. Acad. Sci. U.S.A. 111, 4309–4314. 10.1073/pnas.1317317111 - DOI - PMC - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous