

Delayed Diagnosis and Complications of Predominantly Antibody Deficiencies in a Cohort of Australian Adults
- PMID: 29867917
- PMCID: PMC5960671
- DOI: 10.3389/fimmu.2018.00694
Delayed Diagnosis and Complications of Predominantly Antibody Deficiencies in a Cohort of Australian Adults
Abstract
Background: Predominantly antibody deficiencies (PADs) are the most common type of primary immunodeficiency in adults. PADs frequently pass undetected leading to delayed diagnosis, delayed treatment, and the potential for end-organ damage including bronchiectasis. In addition, PADs are frequently accompanied by comorbid autoimmune disease, and an increased risk of malignancy.
Objectives: To characterize the diagnostic and clinical features of adult PAD patients in Victoria, Australia.
Methods: We identified adult patients receiving, or having previously received immunoglobulin replacement therapy for a PAD at four hospitals in metropolitan Melbourne, and retrospectively characterized their clinical and diagnostic features.
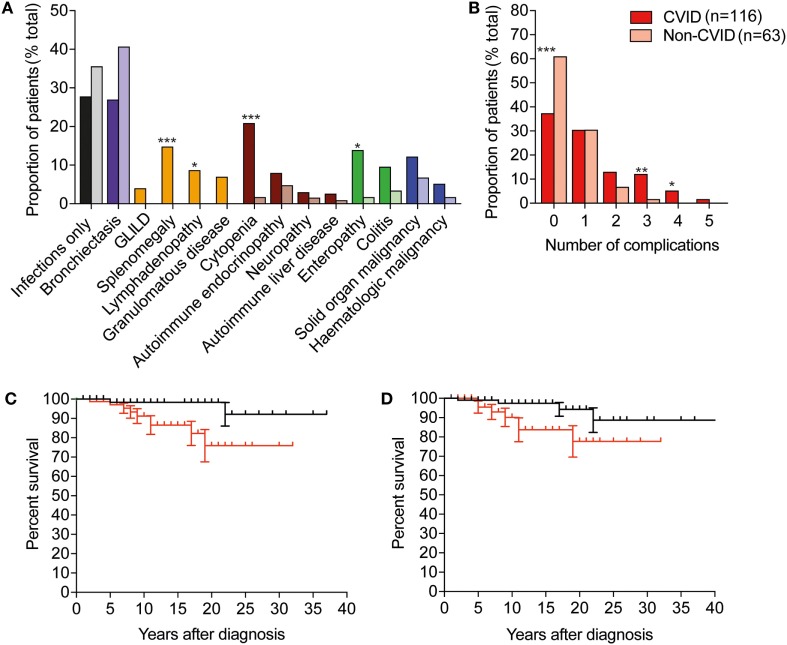
Results: 179 patients from The Royal Melbourne, Alfred and Austin Hospitals, and Monash Medical Centre were included in the study with a median age of 49.7 years (range: 16-87 years), of whom 98 (54.7%) were female. The majority of patients (116; 64.8%) met diagnostic criteria for common variable immunodeficiency (CVID), and 21 (11.7%) were diagnosed with X-linked agammaglobulinemia (XLA). Unclassified hypogammaglobulinemia (HGG) was described in 22 patients (12.3%), IgG subclass deficiency (IGSCD) in 12 (6.7%), and specific antibody deficiency (SpAD) in 4 individuals (2.2%). The remaining four patients had a diagnosis of Good syndrome (thymoma with immunodeficiency). There was no significant difference between the age at diagnosis of the disorders, with the exception of XLA, with a median age at diagnosis of less than 1 year. The median age of reported symptom onset was 20 years for those with a diagnosis of CVID, with a median age at diagnosis of 35 years. CVID patients experienced significantly more non-infectious complications, such as autoimmune cytopenias and lymphoproliferative disease, than the other antibody deficiency disorders. The presence of non-infectious complications was associated with significantly reduced survival in the cohort.
Conclusion: Our data are largely consistent with the experience of other centers internationally, with clear areas for improvement, including reducing diagnostic delay for patients with PADs. It is likely that these challenges will be in part overcome by continued advances in implementation of genomic sequencing for diagnosis of PADs, and with that opportunities for targeted treatment of non-infectious complications.
Keywords: X-linked agammaglobulinemia; common variable immunodeficiency; diagnostic delay; immunoglobulin subclass deficiency; predominantly antibody deficiency; primary immunodeficiency; specific antibody deficiency.
Figures

References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous