

Apert Syndrome With FGFR2 758 C > G Mutation: A Chinese Case Report
- PMID: 29868125
- PMCID: PMC5966571
- DOI: 10.3389/fgene.2018.00181
Apert Syndrome With FGFR2 758 C > G Mutation: A Chinese Case Report
Abstract
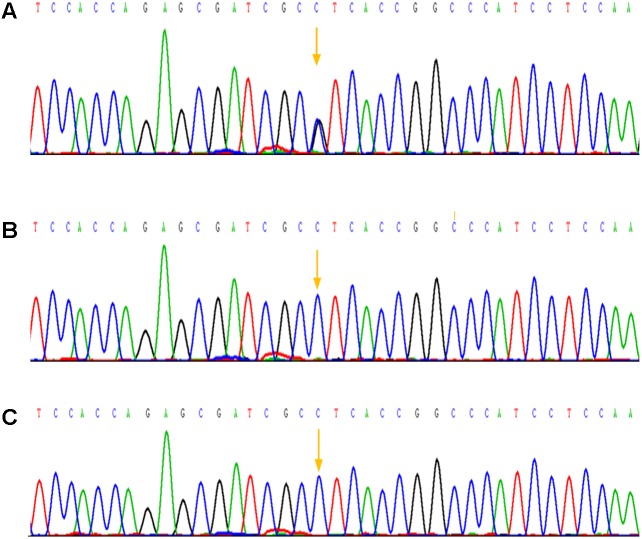
Background: Apert syndrome is considered as one of the most common craniosynostosis syndromes with a prevalence of 1 in 65,000 individuals, and has a close relationship with point mutations in FGFR2 gene. Case report: Here, we described a Apert syndrome case, who was referred to genetic consultation in our hospital with the symptom of craniosynostosis and syndactyly of the hands and feet. Craniosynostosis, midfacial retrusion, steep wide forehead, larger head circumference, marked depression of the nasal bridge, short and wide nose and proptosis could be found obviously, apart from these, ears were mildly low compared with normal children and there was no cleft lip and palate. Mutation was identified by sanger sequencing and a mutation in the exon 7 of FGFR2 gene was detected: p.Pro253Arg (P253R) 758 C > G, which was not found in his parents. Conclusion: The baby had Apert syndrome caused by 758 C > G mutation in the exon 7 of FGFR2 gene, considering no this mutation in his parents, it was spontaneous.
Keywords: Apert syndrome; FGFR2; craniosynostosis; exons sequencing; genetic mutation.
Figures


References
-
- Chang C. C., Tsai F. J., Tsai H. D., Tsai C. H., Hseih Y. Y., Lee C. C., et al. (1998). Prenatal diagnosis of Apert syndrome. 18 621–625. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous