

A comparison of mechanistic signaling pathway activity analysis methods
- PMID: 29868818
- PMCID: PMC6917216
- DOI: 10.1093/bib/bby040
A comparison of mechanistic signaling pathway activity analysis methods
Abstract
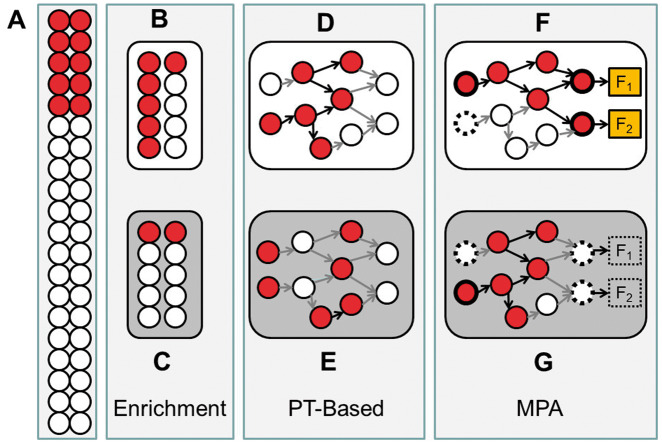
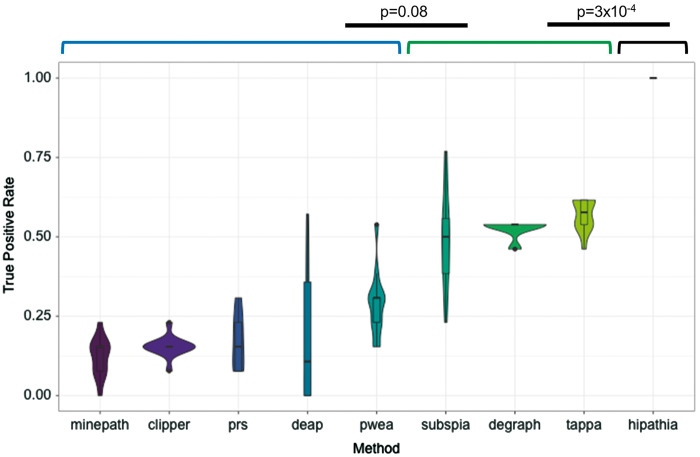
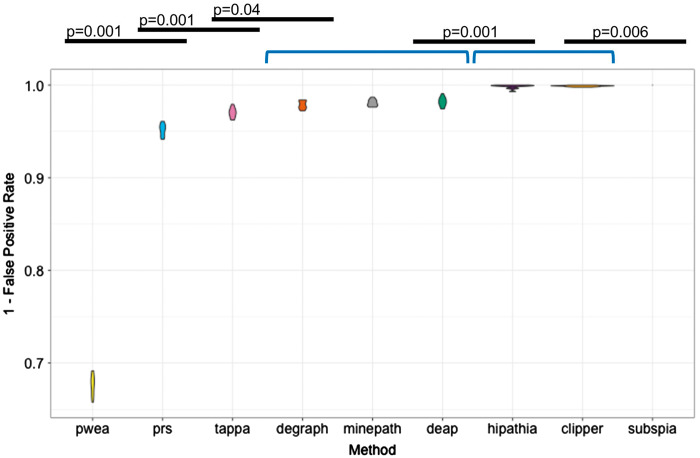
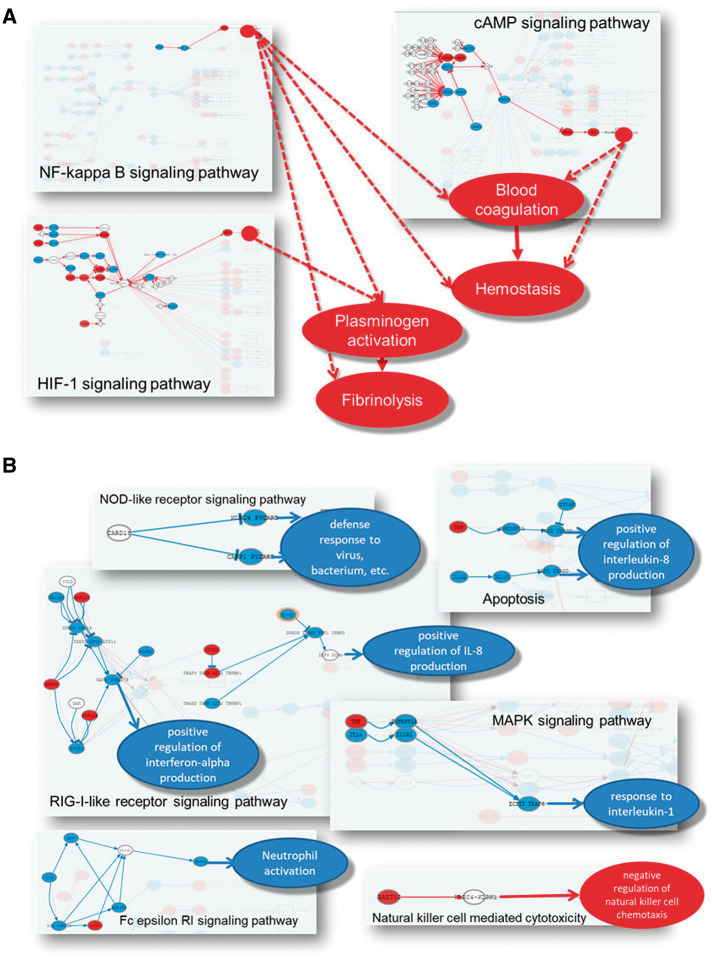
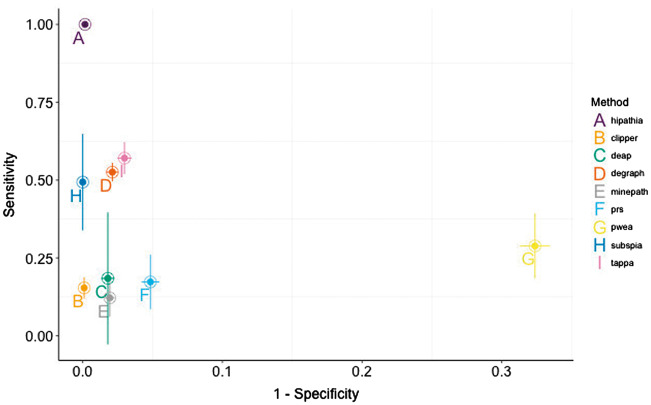
Understanding the aspects of cell functionality that account for disease mechanisms or drug modes of action is a main challenge for precision medicine. Classical gene-based approaches ignore the modular nature of most human traits, whereas conventional pathway enrichment approaches produce only illustrative results of limited practical utility. Recently, a family of new methods has emerged that change the focus from the whole pathways to the definition of elementary subpathways within them that have any mechanistic significance and to the study of their activities. Thus, mechanistic pathway activity (MPA) methods constitute a new paradigm that allows recoding poorly informative genomic measurements into cell activity quantitative values and relate them to phenotypes. Here we provide a review on the MPA methods available and explain their contribution to systems medicine approaches for addressing challenges in the diagnostic and treatment of complex diseases.
Keywords: disease mechanism; mathematical models; networks; signaling pathways; systems biology; transcriptomics.
© The Author(s) 2018. Published by Oxford University Press.
Figures
References
-
- van't Veer LJ, Dai H, van de Vijver MJ, et al.Gene expression profiling predicts clinical outcome of breast cancer. Nature 2002;5(1):530–6. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
