

ISOLDE: a physically realistic environment for model building into low-resolution electron-density maps
- PMID: 29872003
- PMCID: PMC6096486
- DOI: 10.1107/S2059798318002425
ISOLDE: a physically realistic environment for model building into low-resolution electron-density maps
Abstract
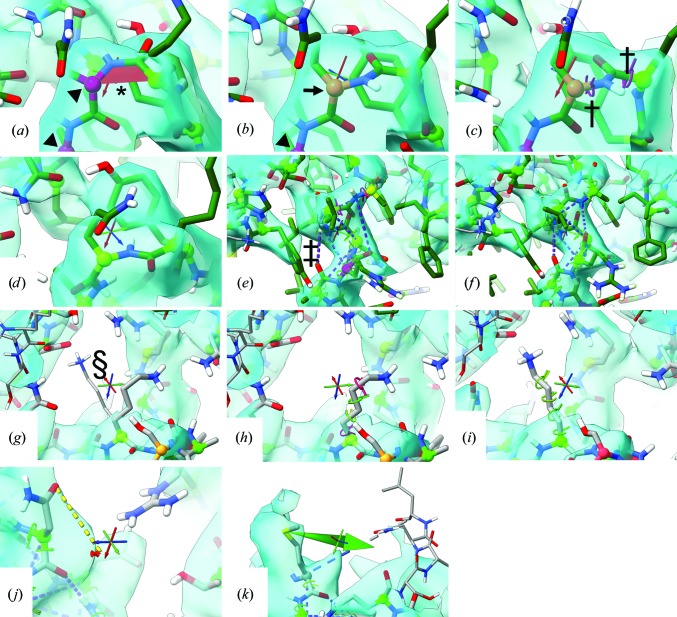
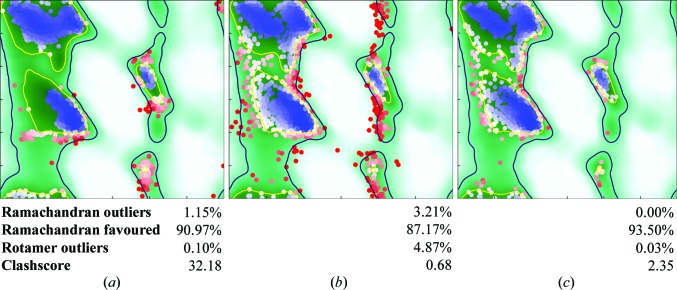
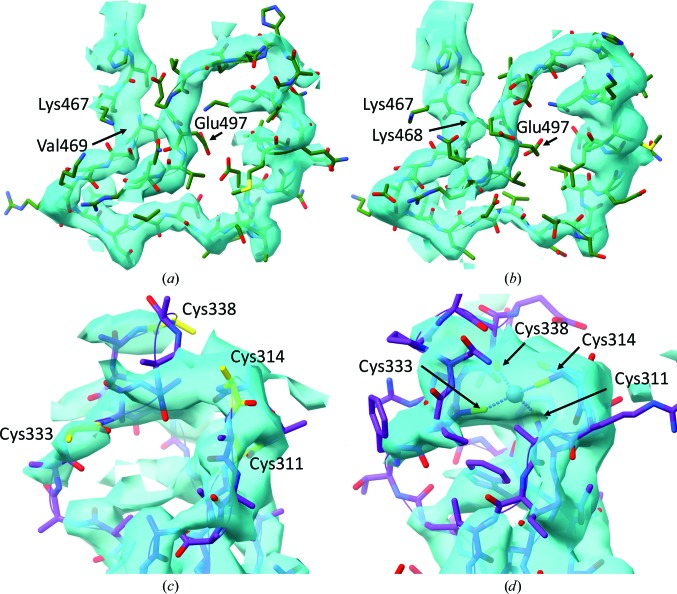
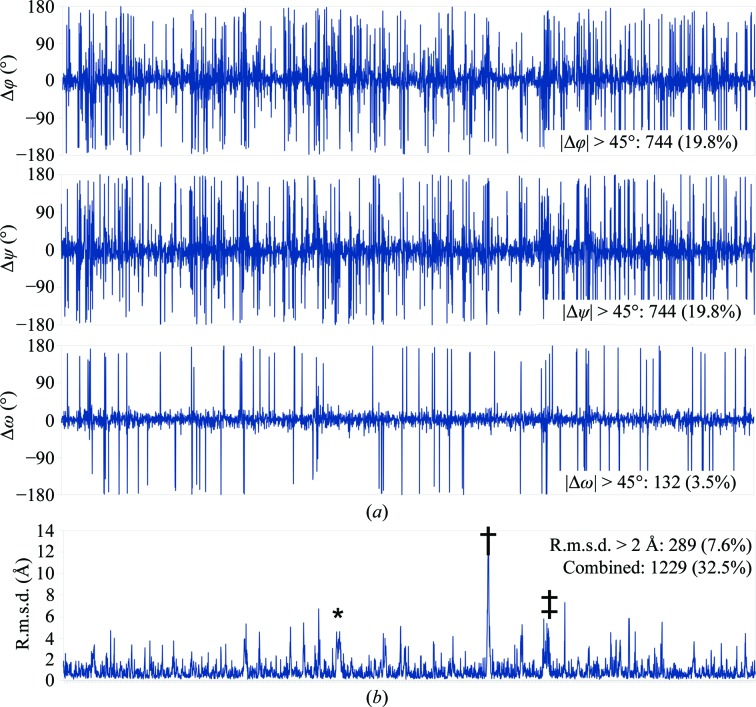
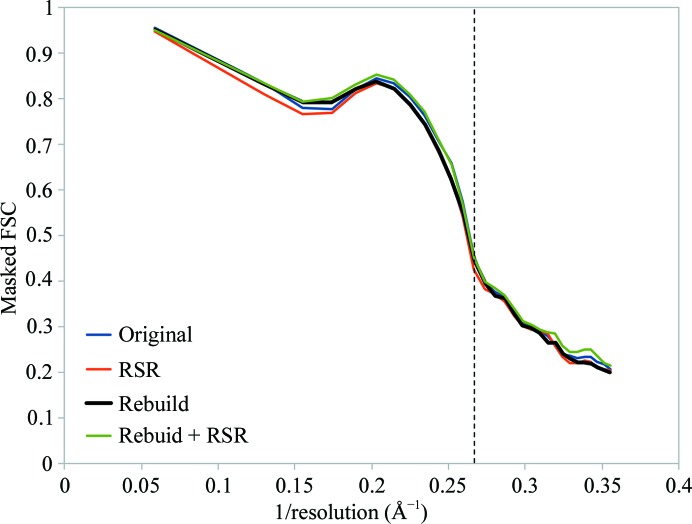
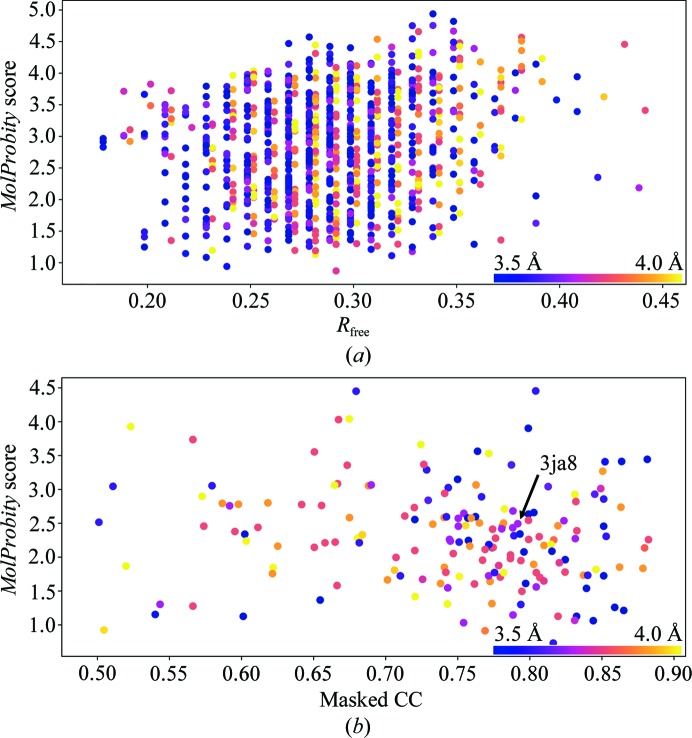
This paper introduces ISOLDE, a new software package designed to provide an intuitive environment for high-fidelity interactive remodelling/refinement of macromolecular models into electron-density maps. ISOLDE combines interactive molecular-dynamics flexible fitting with modern molecular-graphics visualization and established structural biology libraries to provide an immersive interface wherein the model constantly acts to maintain physically realistic conformations as the user interacts with it by directly tugging atoms with a mouse or haptic interface or applying/removing restraints. In addition, common validation tasks are accelerated and visualized in real time. Using the recently described 3.8 Å resolution cryo-EM structure of the eukaryotic minichromosome maintenance (MCM) helicase complex as a case study, it is demonstrated how ISOLDE can be used alongside other modern refinement tools to avoid common pitfalls of low-resolution modelling and improve the quality of the final model. A detailed analysis of changes between the initial and final model provides a somewhat sobering insight into the dangers of relying on a small number of validation metrics to judge the quality of a low-resolution model.
Keywords: ISOLDE; model building; molecular dynamics; real-space refinement; visualization.
open access.
Figures
References
-
- Adams, P. D. et al. (2010). Acta Cryst. D66, 213–221. - PubMed
-
- Afonine, P. V., Klaholz, B. P., Moriarty, N. W., Poon, B. K., Sobolev, O. V., Terwilliger, T. C., Adams, P. D. & Urzhumtsev, A. (2018). bioRxiv, https://doi.org/10.1101/279844.
-
- Brändén, C.-I. & Jones, T. A. (1990). Nature (London), 343, 687–689.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
