

Real-space refinement in PHENIX for cryo-EM and crystallography
- PMID: 29872004
- PMCID: PMC6096492
- DOI: 10.1107/S2059798318006551
Real-space refinement in PHENIX for cryo-EM and crystallography
Abstract
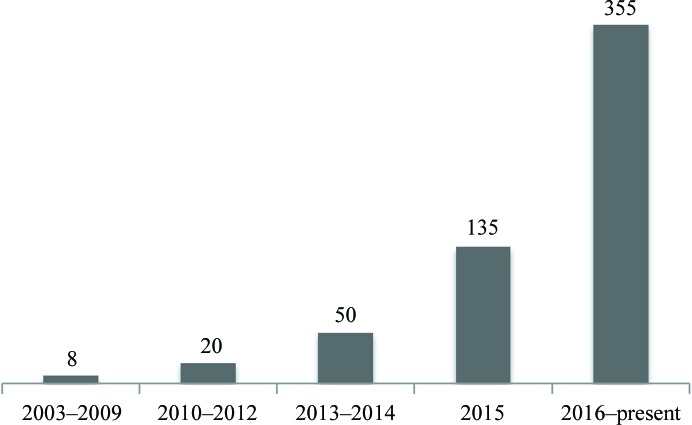
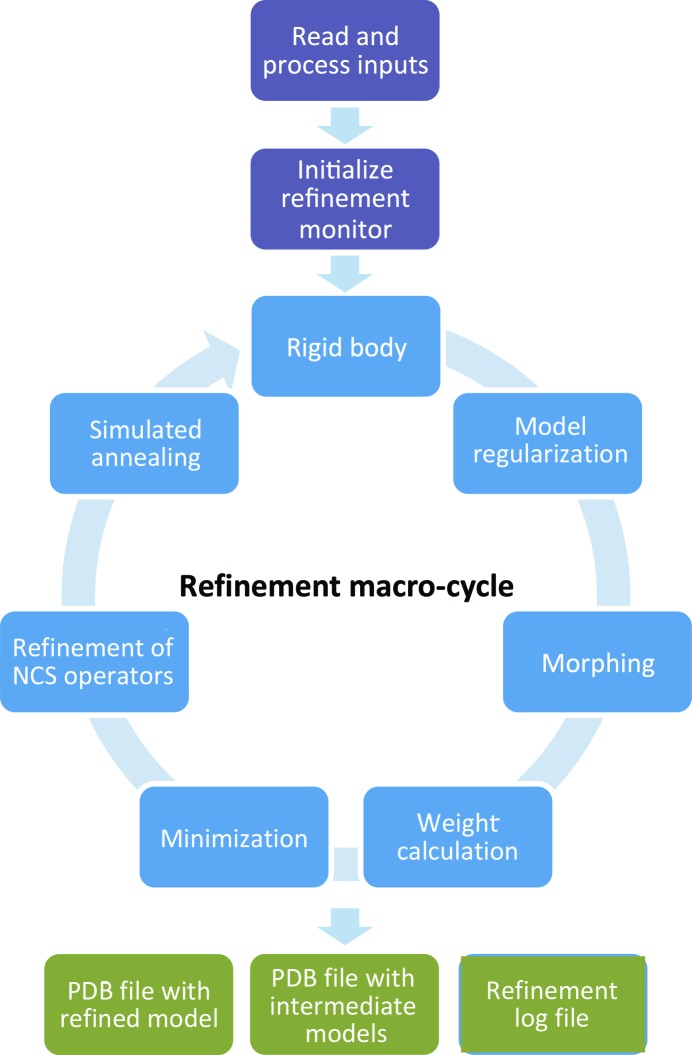
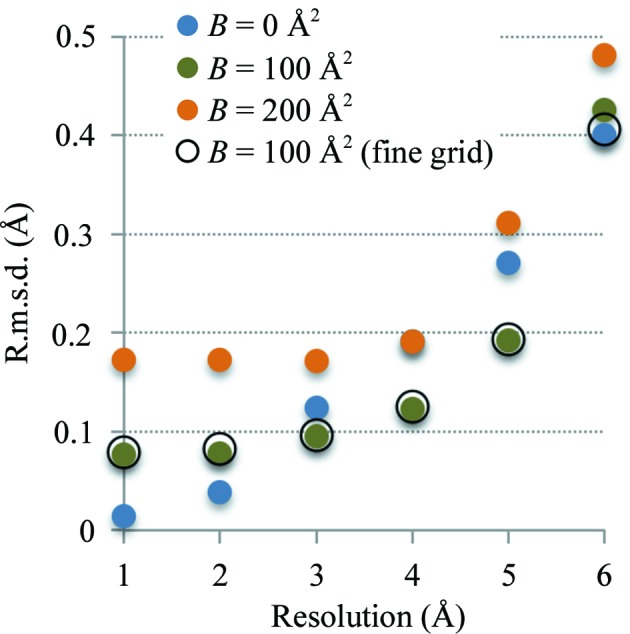
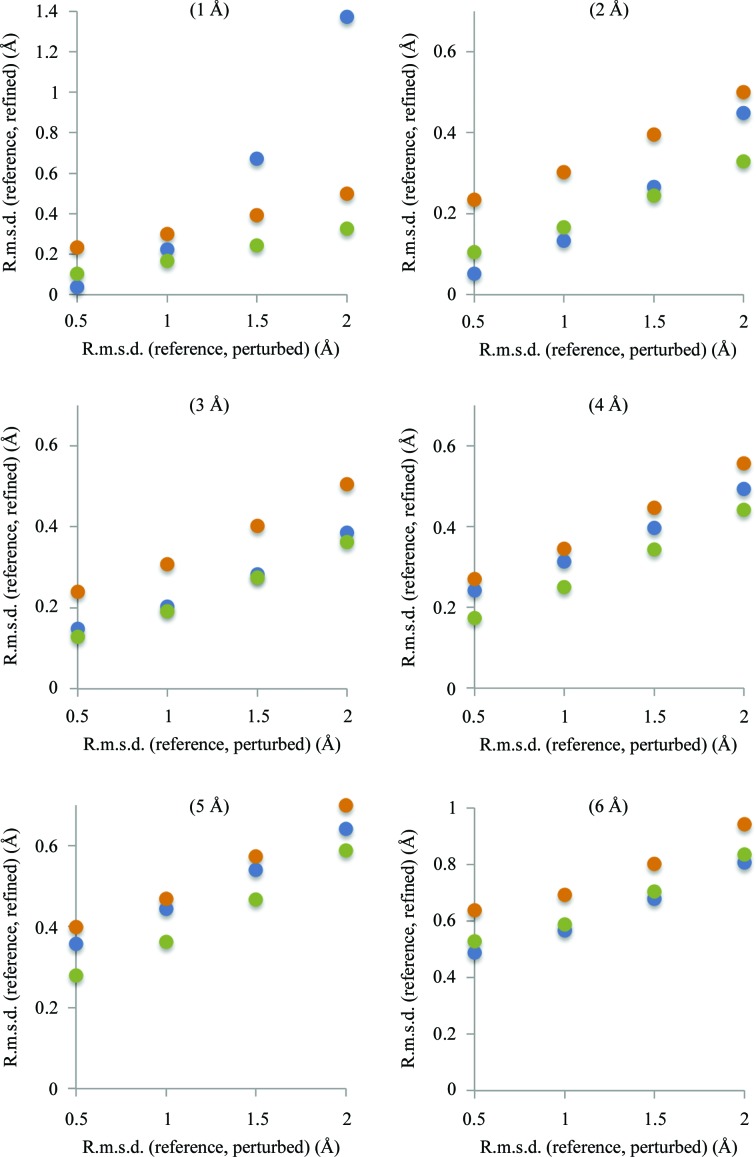
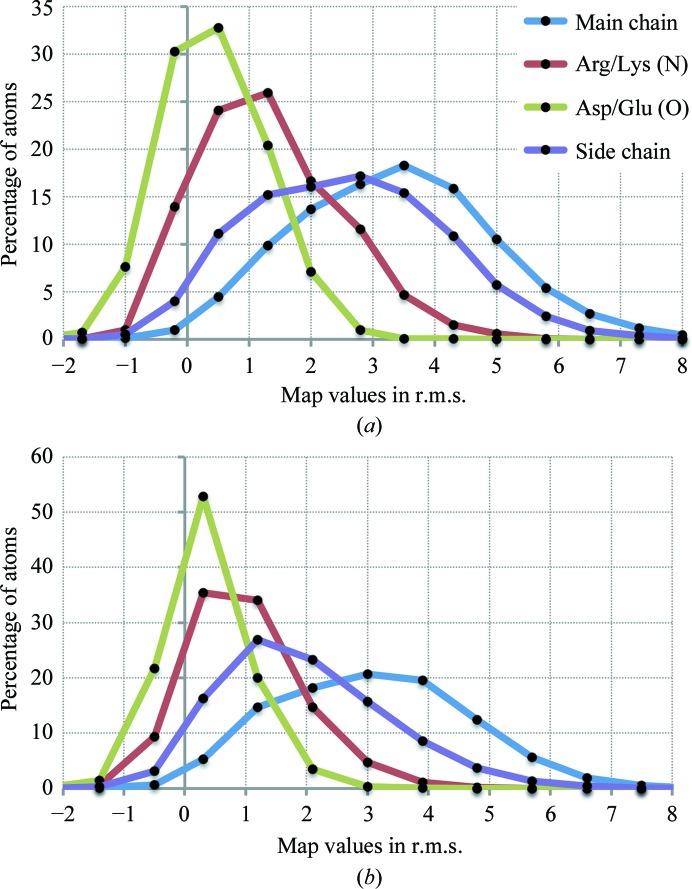
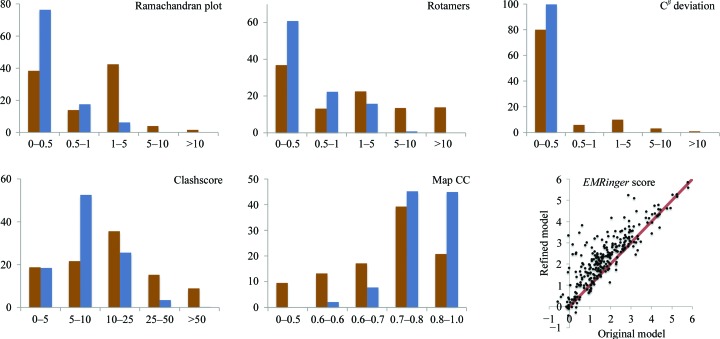
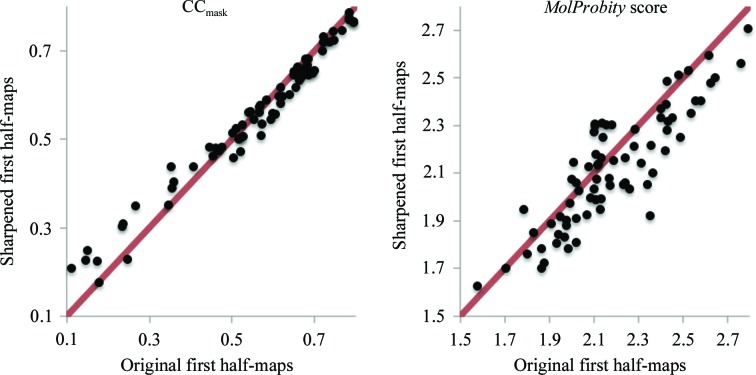
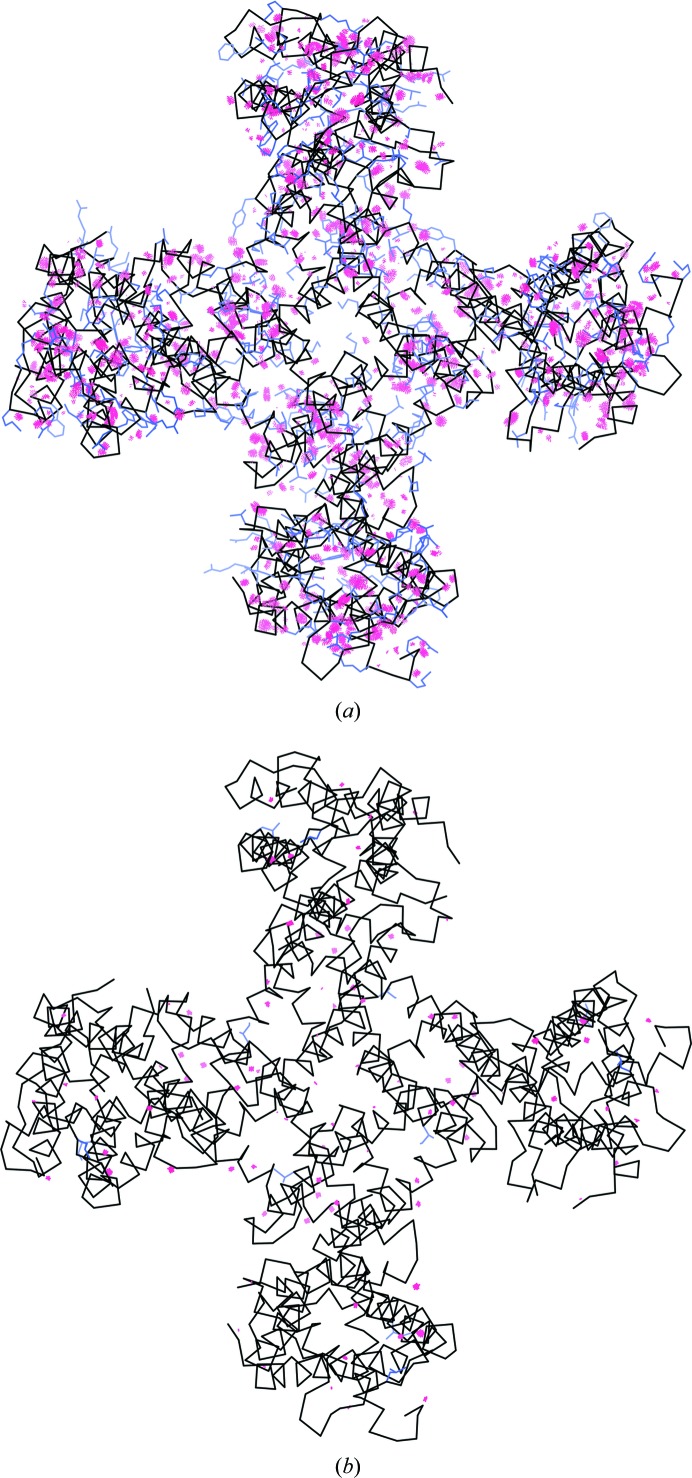
This article describes the implementation of real-space refinement in the phenix.real_space_refine program from the PHENIX suite. The use of a simplified refinement target function enables very fast calculation, which in turn makes it possible to identify optimal data-restraint weights as part of routine refinements with little runtime cost. Refinement of atomic models against low-resolution data benefits from the inclusion of as much additional information as is available. In addition to standard restraints on covalent geometry, phenix.real_space_refine makes use of extra information such as secondary-structure and rotamer-specific restraints, as well as restraints or constraints on internal molecular symmetry. The re-refinement of 385 cryo-EM-derived models available in the Protein Data Bank at resolutions of 6 Å or better shows significant improvement of the models and of the fit of these models to the target maps.
Keywords: PHENIX; atomic-centered targets; cryo-EM; crystallography; map interpolation; real-space refinement.
open access.
Figures
References
-
- Adams, P. D. et al. (2010). Acta Cryst. D66, 213–221.
-
- Afonine, P. V., Headd, J. J., Terwilliger, T. C. & Adams, P. D. (2013). Comput. Crystallogr. Newsl. 4, 43–44. https://www.phenix-online.org/newsletter/CCN_2013_07.pdf.
-
- Afonine, P. V., Klaholz, B. K., Moriarty, N. W., Poon, B. K., Sobolev, O. V., Terwilliger, T. C., Adams, P. D. & Urzhumtsev, A. (2018). bioRxiv. https://doi.org/10.1101/249607.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
