

Interaction between Wnt/β-catenin and RAS-ERK pathways and an anti-cancer strategy via degradations of β-catenin and RAS by targeting the Wnt/β-catenin pathway
- PMID: 29872723
- PMCID: PMC5871897
- DOI: 10.1038/s41698-018-0049-y
Interaction between Wnt/β-catenin and RAS-ERK pathways and an anti-cancer strategy via degradations of β-catenin and RAS by targeting the Wnt/β-catenin pathway
Abstract
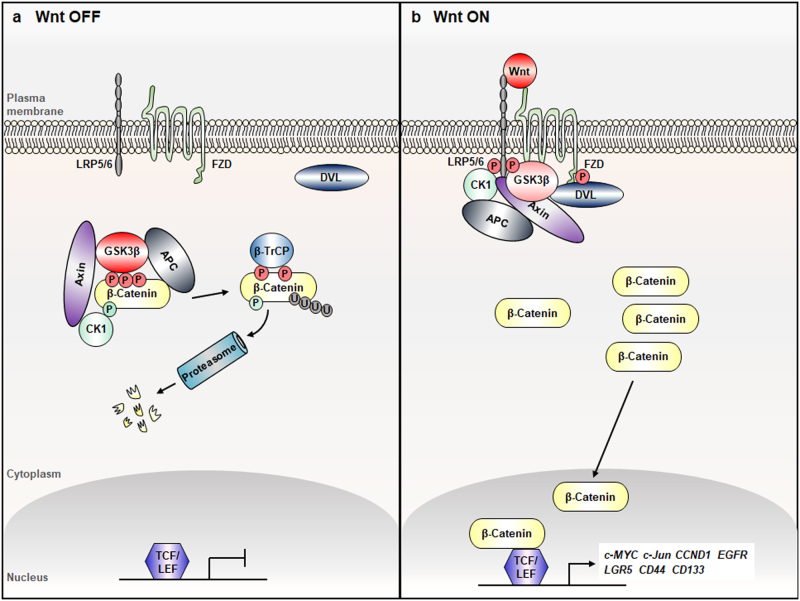
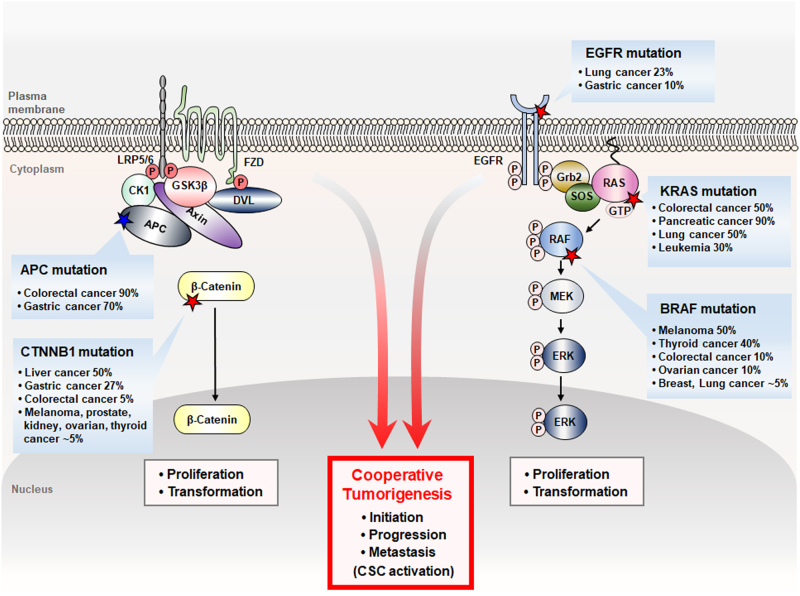
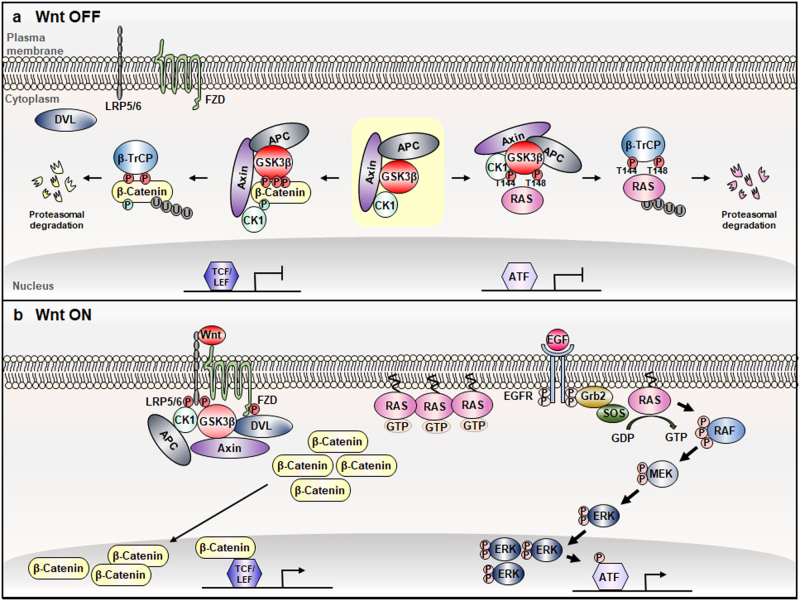
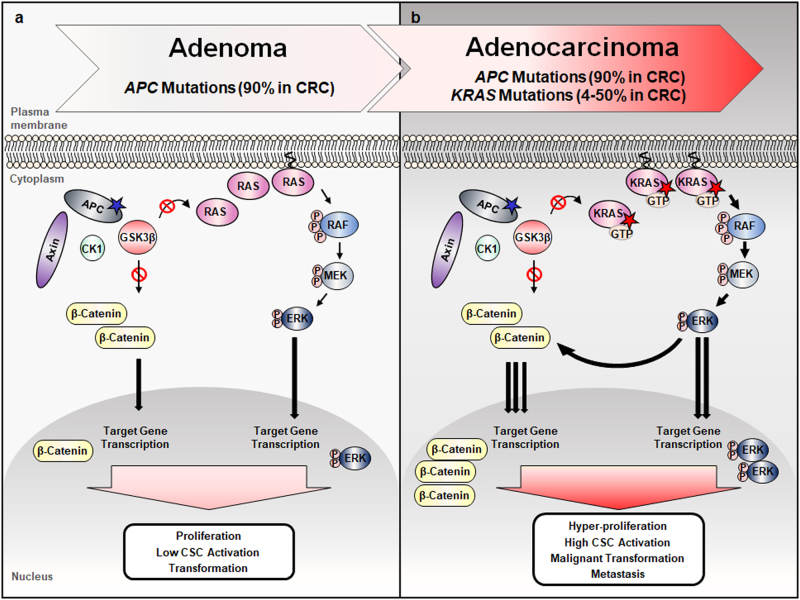
Aberrant activation of the Wnt/β-catenin and RAS-extracellular signal-regulated kinase (ERK) pathways play important roles in the tumorigenesis of many different types of cancer, most notably colorectal cancer (CRC). Genes for these two pathways, such as adenomatous polyposis coli (APC) and KRAS are frequently mutated in human CRC, and involved in the initiation and progression of the tumorigenesis, respectively. Moreover, recent studies revealed interaction of APC and KRAS mutations in the various stages of colorectal tumorigenesis and even in metastasis accompanying activation of the cancer stem cells (CSCs). A key event in the synergistic cooperation between Wnt/β-catenin and RAS-ERK pathways is a stabilization of both β-catenin and RAS especially mutant KRAS by APC loss, and pathological significance of this was indicated by correlation of increased β-catenin and RAS levels in human CRC where APC mutations occur as high as 90% of CRC patients. Together with the notion of the protein activity reduction by lowering its level, inhibition of both β-catenin and RAS especially by degradation could be a new ideal strategy for development of anti-cancer drugs for CRC. In this review, we will discuss interaction between the Wnt/β-catenin and RAS-ERK pathways in the colorectal tumorigenesis by providing the mechanism of RAS stabilization by aberrant activation of Wnt/β-catenin. We will also discuss our small molecular anti-cancer approach controlling CRC by induction of specific degradations of both β-catenin and RAS via targeting Wnt/β-catenin pathway especially for the KYA1797K, a small molecule specifically binding at the regulator of G-protein signaling (RGS)-domain of Axin.
Conflict of interest statement
The authors declare no competing financial interests.
Figures

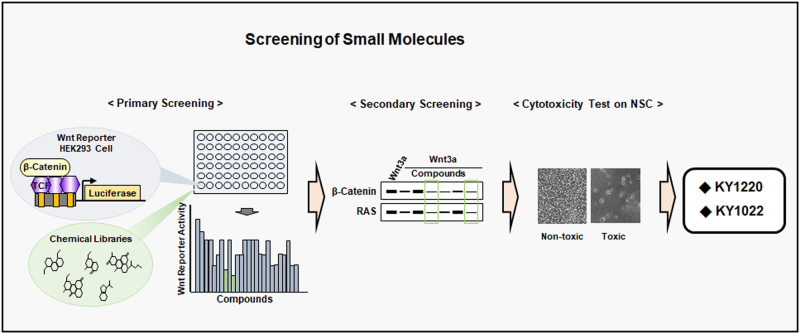
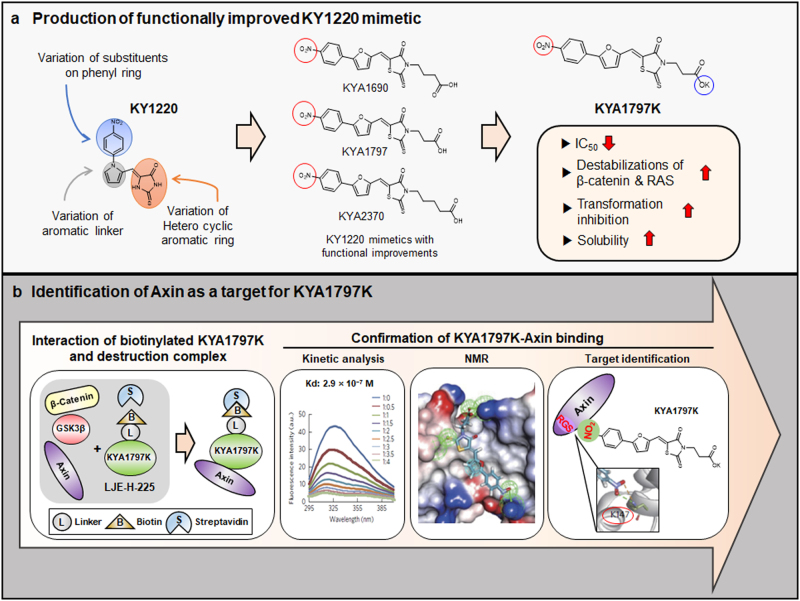
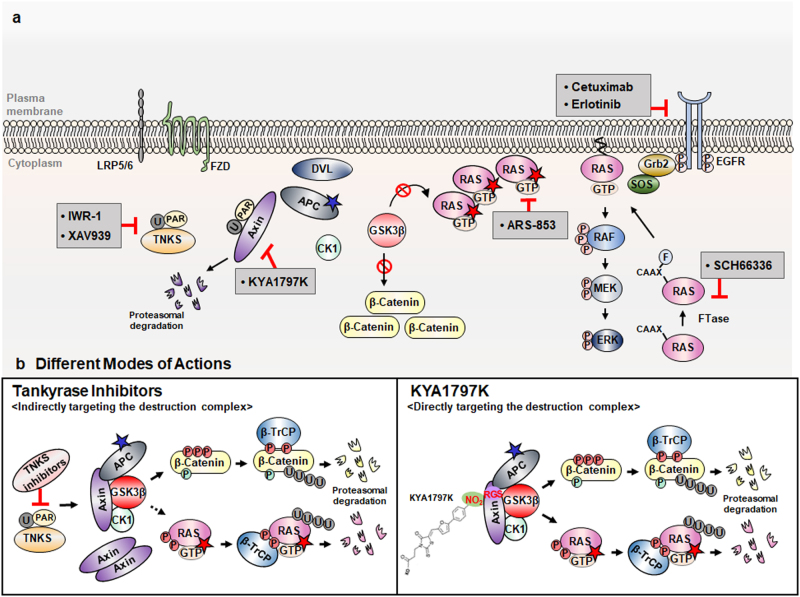
References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
