

Rosetta Protein Structure Prediction from Hydroxyl Radical Protein Footprinting Mass Spectrometry Data
- PMID: 29874044
- PMCID: PMC6008241
- DOI: 10.1021/acs.analchem.8b01624
Rosetta Protein Structure Prediction from Hydroxyl Radical Protein Footprinting Mass Spectrometry Data
Abstract
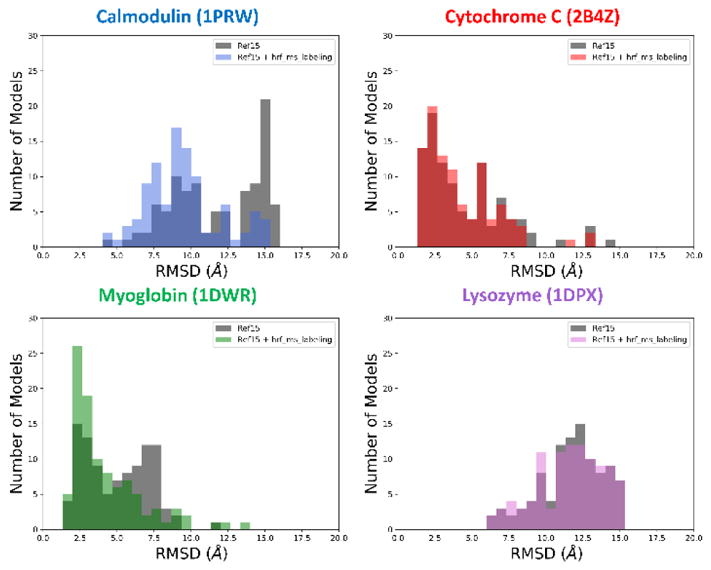
In recent years mass spectrometry-based covalent labeling techniques such as hydroxyl radical footprinting (HRF) have emerged as valuable structural biology techniques, yielding information on protein tertiary structure. These data, however, are not sufficient to predict protein structure unambiguously, as they provide information only on the relative solvent exposure of certain residues. Despite some recent advances, no software currently exists that can utilize covalent labeling mass spectrometry data to predict protein tertiary structure. We have developed the first such tool, which incorporates mass spectrometry derived protection factors from HRF labeling as a new centroid score term for the Rosetta scoring function to improve the prediction of protein tertiary structures. We tested our method on a set of four soluble benchmark proteins with known crystal structures and either published HRF experimental results or internally acquired data. Using the HRF labeling data, we rescored large decoy sets of structures predicted with Rosetta for each of the four benchmark proteins. As a result, the model quality improved for all benchmark proteins as compared to when scored with Rosetta alone. For two of the four proteins we were even able to identify atomic resolution models with the addition of HRF data.
Figures




References
-
- Fenn JB, Mann M, Meng CK, Wong SF, Whitehouse CM. Electrospray ionization for mass spectrometry of large biomolecules. Science. 1989;246(4926):64–71. - PubMed
-
- Link AJ, Eng J, Schieltz DM, Carmack E, Mize GJ, Morris DR, Garvik BM, Yates JR., III Direct analysis of protein complexes using mass spectrometry. Nature Biotechnology. 1999;17(7):676. - PubMed
-
- Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature. 2003 - PubMed
-
- Küster B, Mann M. Identifying proteins and post-translational modifications by mass spectrometry. Current Opinion in Structural Biology. 1998;8(3):393–400. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
