

Niemann-Pick Type C2 Protein Regulates Free Cholesterol Accumulation and Influences Hepatic Stellate Cell Proliferation and Mitochondrial Respiration Function
- PMID: 29874879
- PMCID: PMC6032364
- DOI: 10.3390/ijms19061678
Niemann-Pick Type C2 Protein Regulates Free Cholesterol Accumulation and Influences Hepatic Stellate Cell Proliferation and Mitochondrial Respiration Function
Abstract
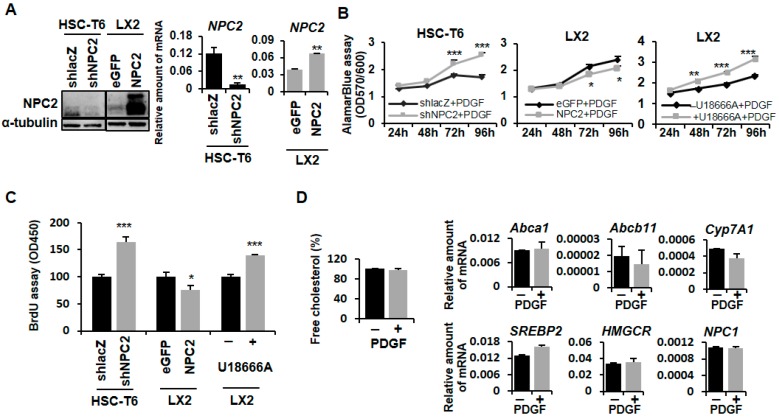
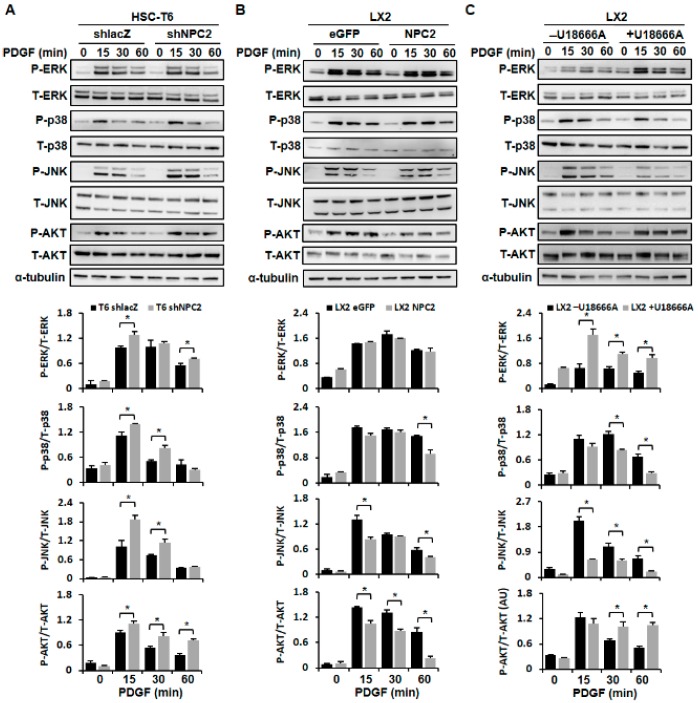
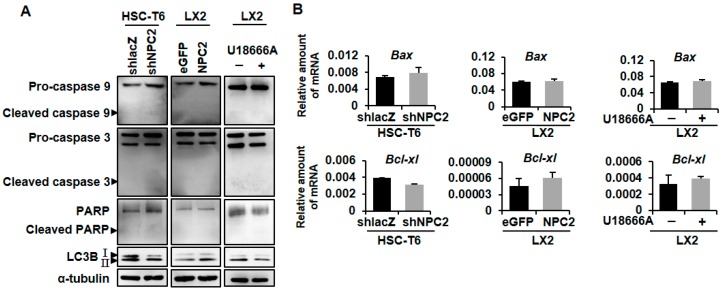
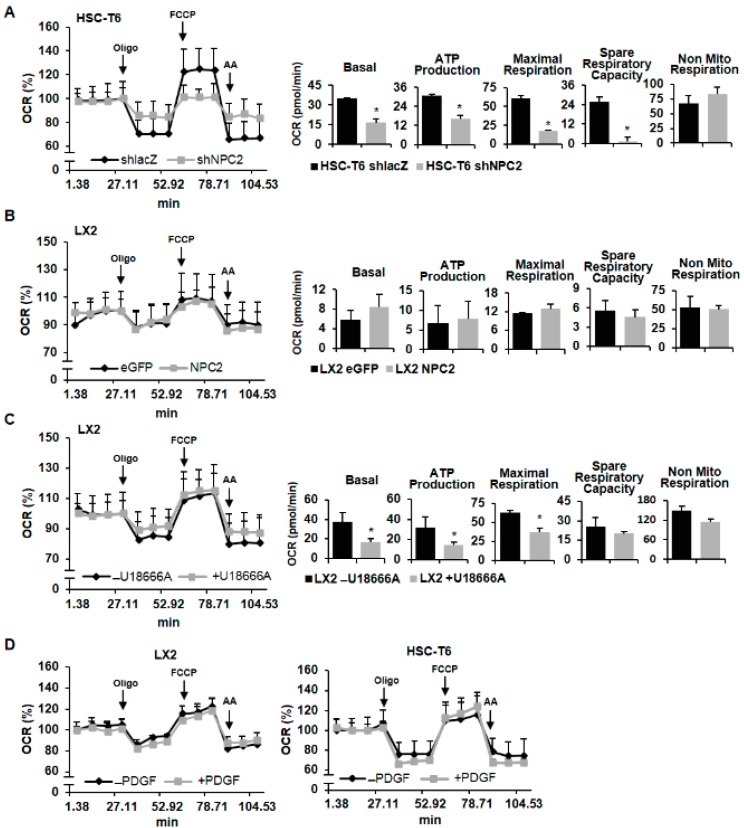
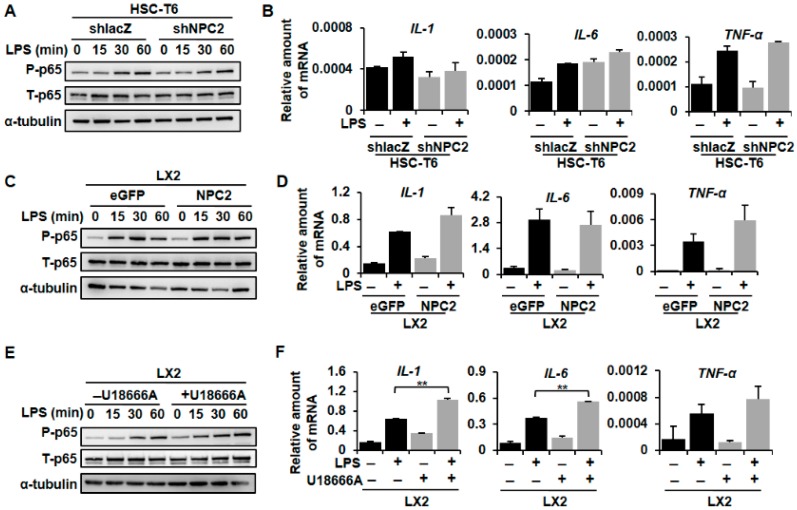
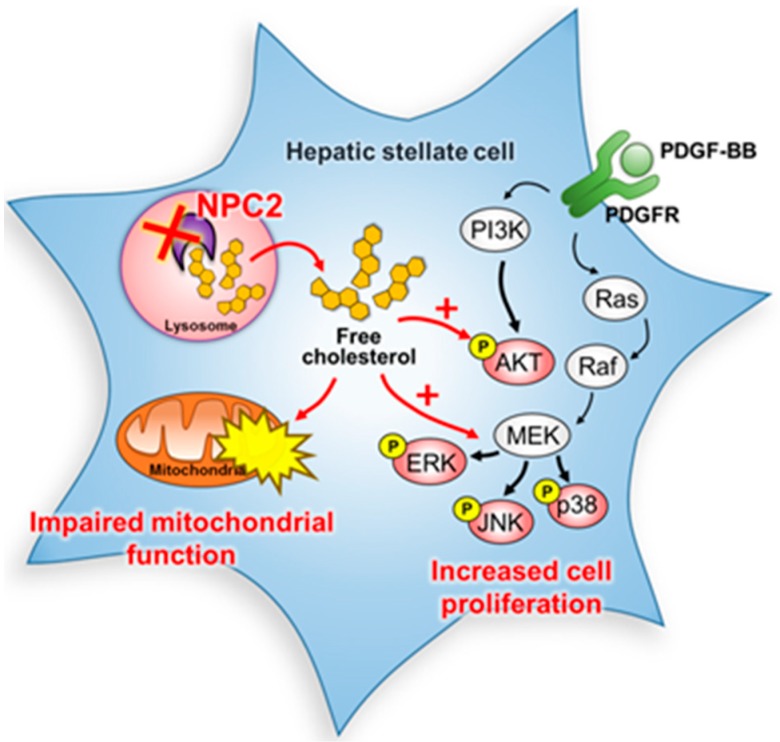
Liver fibrosis is the first step toward the progression to cirrhosis, portal hypertension, and hepatocellular carcinoma. A high-cholesterol diet is associated with liver fibrosis via the accumulation of free cholesterol in hepatic stellate cells (HSCs). Niemann-Pick type C2 (NPC2) plays an important role in the regulation of intracellular free cholesterol homeostasis via direct binding with free cholesterol. Previously, we reported that NPC2 was downregulated in liver cirrhosis tissues. Loss of NPC2 enhanced the accumulation of free cholesterol in HSCs and made them more susceptible to transforming growth factor (TGF)-β1. In this study, we showed that knockdown of NPC2 resulted in marked increases in platelet-derived growth factor BB (PDGF-BB)-induced HSC proliferation through enhanced extracellular signal-regulated kinases (ERK), p38, c-Jun N-terminal kinases (JNK), and protein kinase B (AKT) phosphorylation. In contrast, NPC2 overexpression decreased PDGF-BB-induced cell proliferation by inhibiting p38, JNK, and AKT phosphorylation. Although NPC2 expression did not affect caspase-related apoptosis, the autophagy marker light chain 3β (LC3B) was decreased in NPC2 knockdown, and free cholesterol accumulated in the HSCs. The mitochondrial respiration functions (such as oxygen consumption rate, ATP production, and maximal respiratory capacity) were decreased in NPC2 knockdown, and free cholesterol accumulated in the HSCs, while NPC2-overexpressed cells remained normal. In addition, NPC2 expression did not affect the susceptibility of HSCs to lipopolysaccharides (LPS), and U18666A treatment induced free cholesterol accumulation, which enhanced LPS-induced Toll-like receptor 4 (TLR4), nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) p65 phosphorylation, interleukin (IL)-1 and IL-6 expression. Our study demonstrated that NPC2-mediated free cholesterol homeostasis controls HSC proliferation and mitochondrial function.
Keywords: Niemann-Pick type C2; free cholesterol; hepatic stellate cells; mitochondrial function; platelet-derived growth factor BB.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

References
-
- Moreira R.K. Hepatic stellate cells and liver fibrosis. Arch. Pathol. Lab. Med. 2007;131:1728–1734. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
