

Smooth muscle glucose metabolism promotes monocyte recruitment and atherosclerosis in a mouse model of metabolic syndrome
- PMID: 29875324
- PMCID: PMC6124428
- DOI: 10.1172/jci.insight.96544
Smooth muscle glucose metabolism promotes monocyte recruitment and atherosclerosis in a mouse model of metabolic syndrome
Abstract
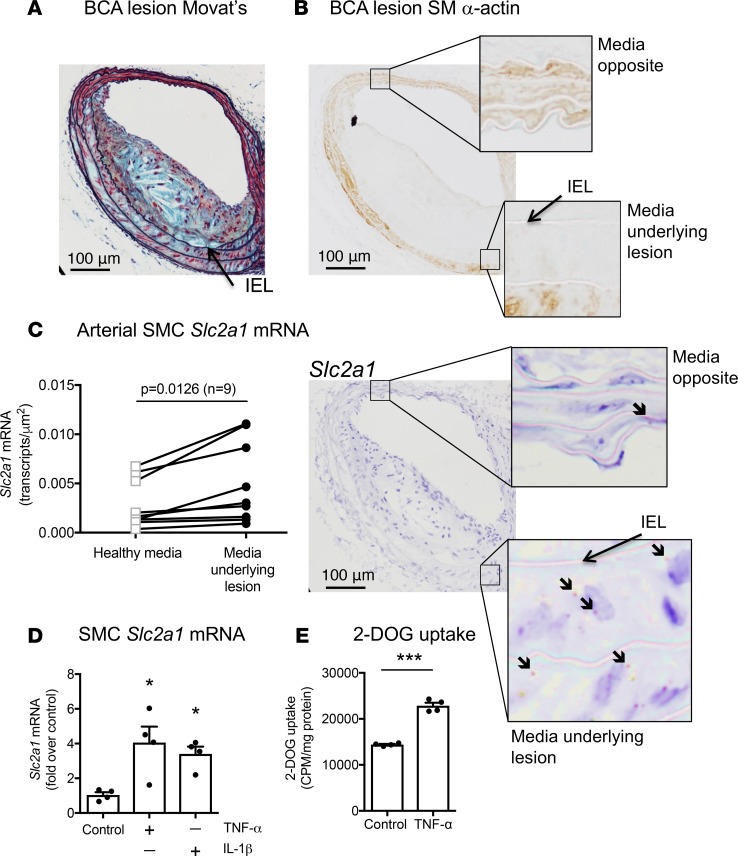
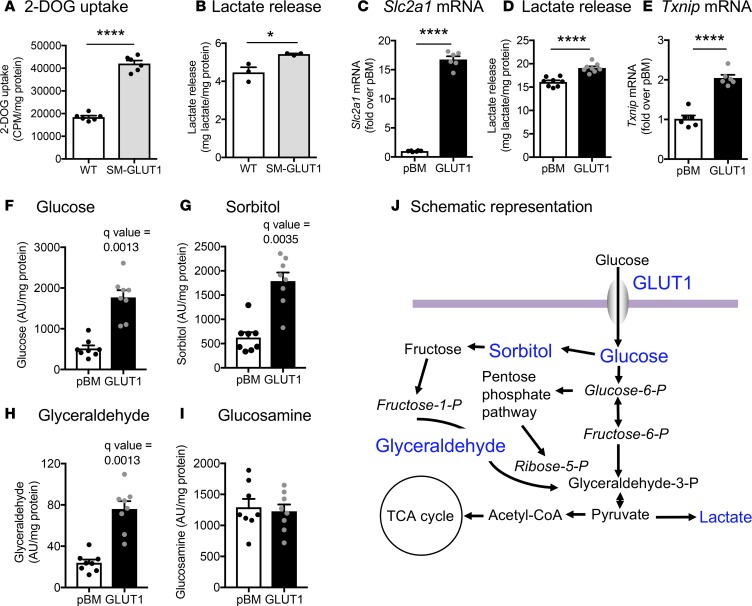
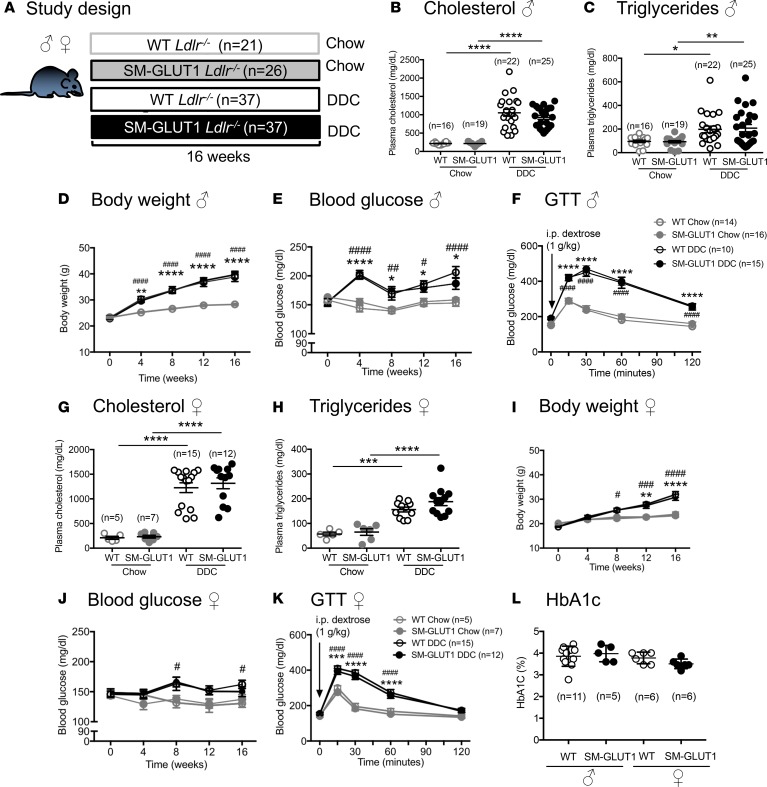
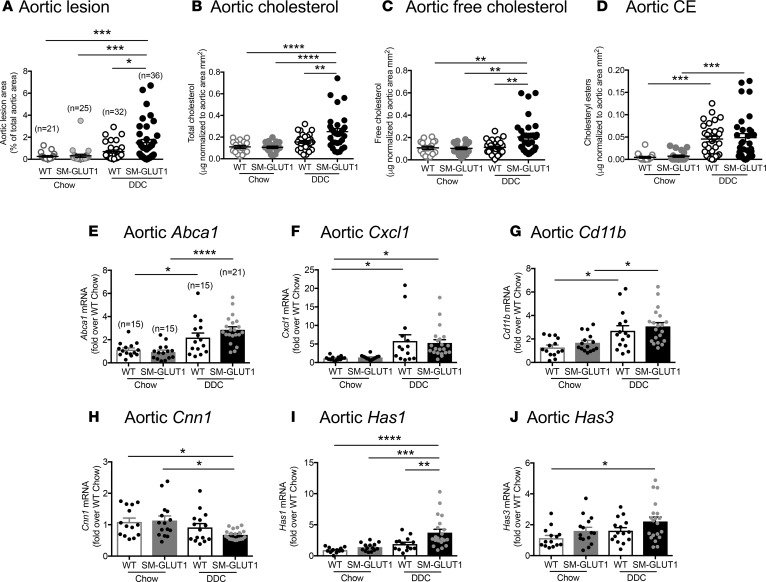
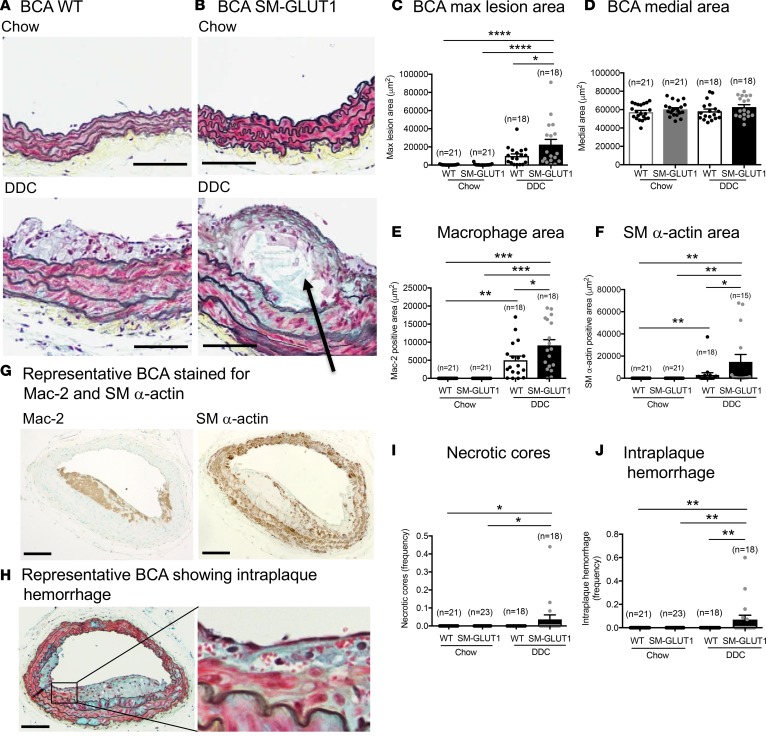
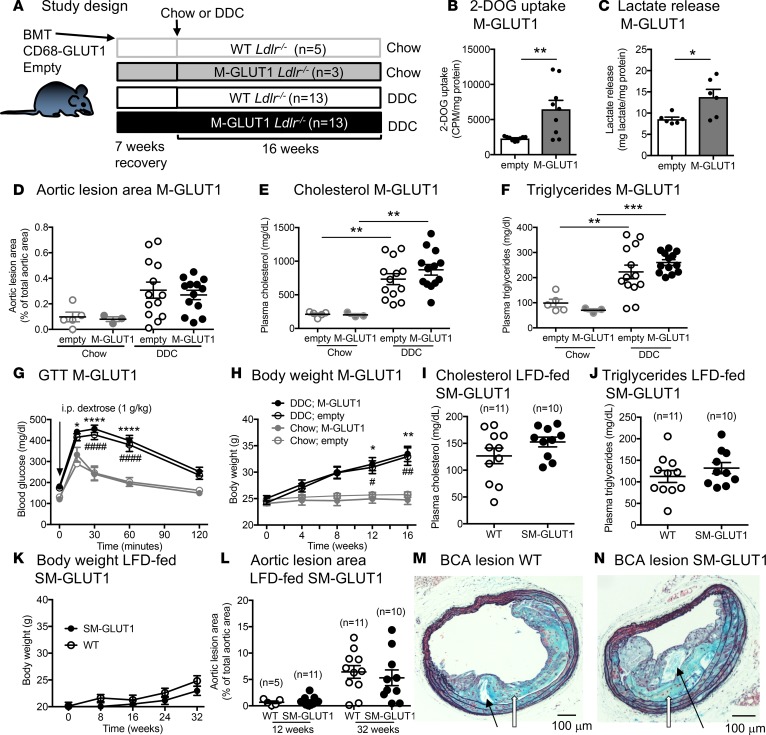
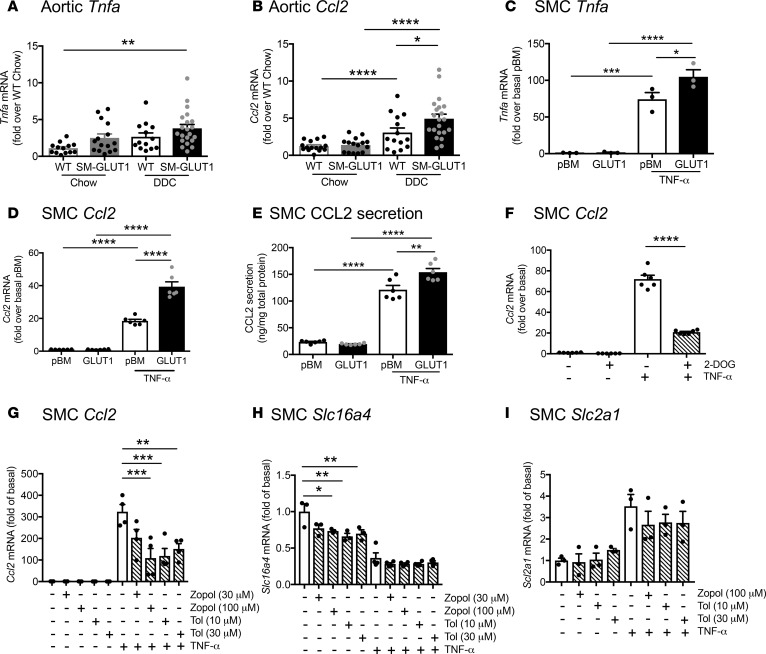
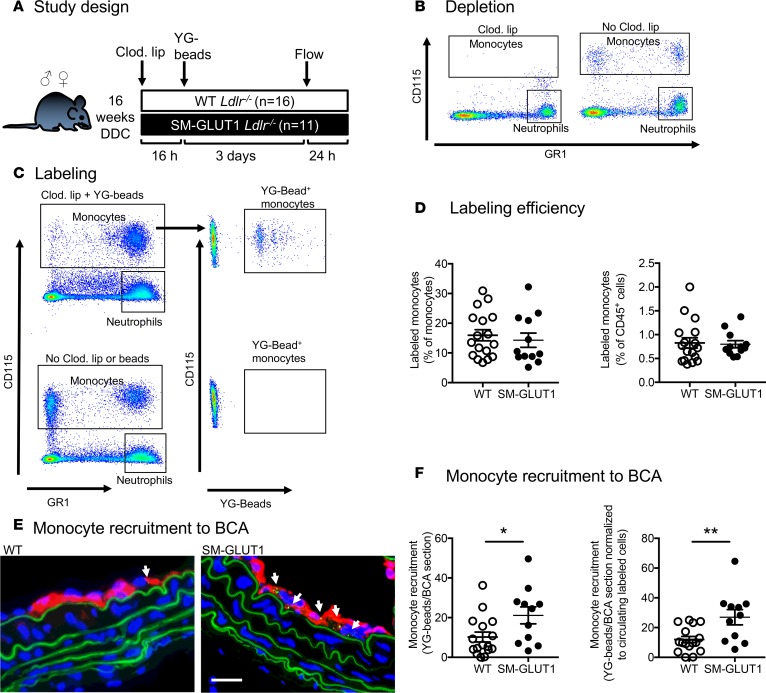
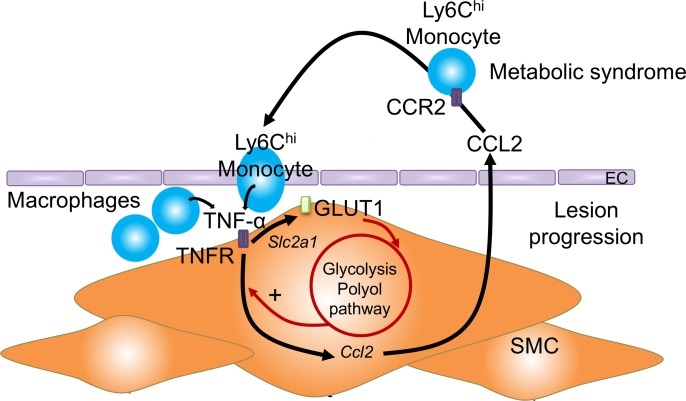
Metabolic syndrome contributes to cardiovascular disease partly through systemic risk factors. However, local processes in the artery wall are becoming increasingly recognized to exacerbate atherosclerosis both in mice and humans. We show that arterial smooth muscle cell (SMC) glucose metabolism markedly synergizes with metabolic syndrome in accelerating atherosclerosis progression, using a low-density lipoprotein receptor-deficient mouse model. SMCs in proximity to atherosclerotic lesions express increased levels of the glucose transporter GLUT1. Cytokines, such as TNF-α produced by lesioned arteries, promote GLUT1 expression in SMCs, which in turn increases expression of the chemokine CCL2 through increased glycolysis and the polyol pathway. Furthermore, overexpression of GLUT1 in SMCs, but not in myeloid cells, accelerates development of larger, more advanced lesions in a mouse model of metabolic syndrome, which also exhibits elevated levels of circulating Ly6Chi monocytes expressing the CCL2 receptor CCR2. Accordingly, monocyte tracing experiments demonstrate that targeted SMC GLUT1 overexpression promotes Ly6Chi monocyte recruitment to lesions. Strikingly, SMC-targeted GLUT1 overexpression fails to accelerate atherosclerosis in mice that do not exhibit the metabolic syndrome phenotype or monocytosis. These results reveal a potentially novel mechanism whereby arterial smooth muscle glucose metabolism synergizes with metabolic syndrome to accelerate monocyte recruitment and atherosclerosis progression.
Keywords: Atherosclerosis; Glucose metabolism; Vascular Biology.
Conflict of interest statement
Figures
Similar articles
-
CD8+ T Cells Drive Plaque Smooth Muscle Cell Dedifferentiation in Experimental Atherosclerosis.Arterioscler Thromb Vasc Biol. 2024 Aug;44(8):1852-1872. doi: 10.1161/ATVBAHA.123.320084. Epub 2024 Jun 13. Arterioscler Thromb Vasc Biol. 2024. PMID: 38868941
-
Translatome profiling reveals Itih4 as a novel smooth muscle cell-specific gene in atherosclerosis.Cardiovasc Res. 2024 Jul 2;120(8):869-882. doi: 10.1093/cvr/cvae028. Cardiovasc Res. 2024. PMID: 38289873 Free PMC article.
-
Aldosterone increases early atherosclerosis and promotes plaque inflammation through a placental growth factor-dependent mechanism.J Am Heart Assoc. 2013 Feb 22;2(1):e000018. doi: 10.1161/JAHA.112.000018. J Am Heart Assoc. 2013. PMID: 23525413 Free PMC article.
-
Contribution of monocyte-derived macrophages and smooth muscle cells to arterial foam cell formation.Cardiovasc Res. 2012 Jul 15;95(2):165-72. doi: 10.1093/cvr/cvs094. Epub 2012 Feb 15. Cardiovasc Res. 2012. PMID: 22345306 Review.
-
Cholesterol homeostasis and high-density lipoprotein formation in arterial smooth muscle cells.Trends Cardiovasc Med. 2010 Apr;20(3):96-102. doi: 10.1016/j.tcm.2010.09.002. Trends Cardiovasc Med. 2010. PMID: 21130953 Review.
Cited by
-
VNS improves VSMC metabolism and arteriogenesis in infarcted hearts through m/n-AChR-Akt-SDF-1α in adult male rats.J Mol Histol. 2024 Feb;55(1):51-67. doi: 10.1007/s10735-023-10171-4. Epub 2024 Jan 2. J Mol Histol. 2024. PMID: 38165566 Free PMC article.
-
Analysis of Gene Expression Profiles in the Liver of Rats With Intrauterine Growth Retardation.Front Pediatr. 2022 Mar 7;10:801544. doi: 10.3389/fped.2022.801544. eCollection 2022. Front Pediatr. 2022. PMID: 35321016 Free PMC article.
-
GPR40 deficiency worsens metabolic syndrome-associated periodontitis in mice.J Periodontal Res. 2023 Jun;58(3):575-587. doi: 10.1111/jre.13107. Epub 2023 Feb 17. J Periodontal Res. 2023. PMID: 36807310 Free PMC article.
-
Insulin receptors in vascular smooth muscle cells regulate plaque stability of atherosclerosis.Cardiovasc Res. 2024 Dec 14;120(16):2017-2030. doi: 10.1093/cvr/cvae193. Cardiovasc Res. 2024. PMID: 39197028 Free PMC article.
-
Obese mice have decreased uterine contractility and altered energy metabolism in the uterus at term gestation†.Biol Reprod. 2024 Sep 14;111(3):678-693. doi: 10.1093/biolre/ioae086. Biol Reprod. 2024. PMID: 38857377 Free PMC article.
References
-
- [No authors listed] Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S) Lancet. 1994;344(8934):1383–1389. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
