

Aminoacyl-tRNA synthetase deficiencies in search of common themes
- PMID: 29875423
- PMCID: PMC7091658
- DOI: 10.1038/s41436-018-0048-y
Aminoacyl-tRNA synthetase deficiencies in search of common themes
Erratum in
-
Correction: Aminoacyl-tRNA synthetase deficiencies in search of common themes.Genet Med. 2021 Oct;23(10):2024. doi: 10.1038/s41436-020-00966-1. Genet Med. 2021. PMID: 32934367 Free PMC article. No abstract available.
Abstract
Purpose: Pathogenic variations in genes encoding aminoacyl-tRNA synthetases (ARSs) are increasingly associated with human disease. Clinical features of autosomal recessive ARS deficiencies appear very diverse and without apparent logic. We searched for common clinical patterns to improve disease recognition, insight into pathophysiology, and clinical care.
Methods: Symptoms were analyzed in all patients with recessive ARS deficiencies reported in literature, supplemented with unreported patients evaluated in our hospital.
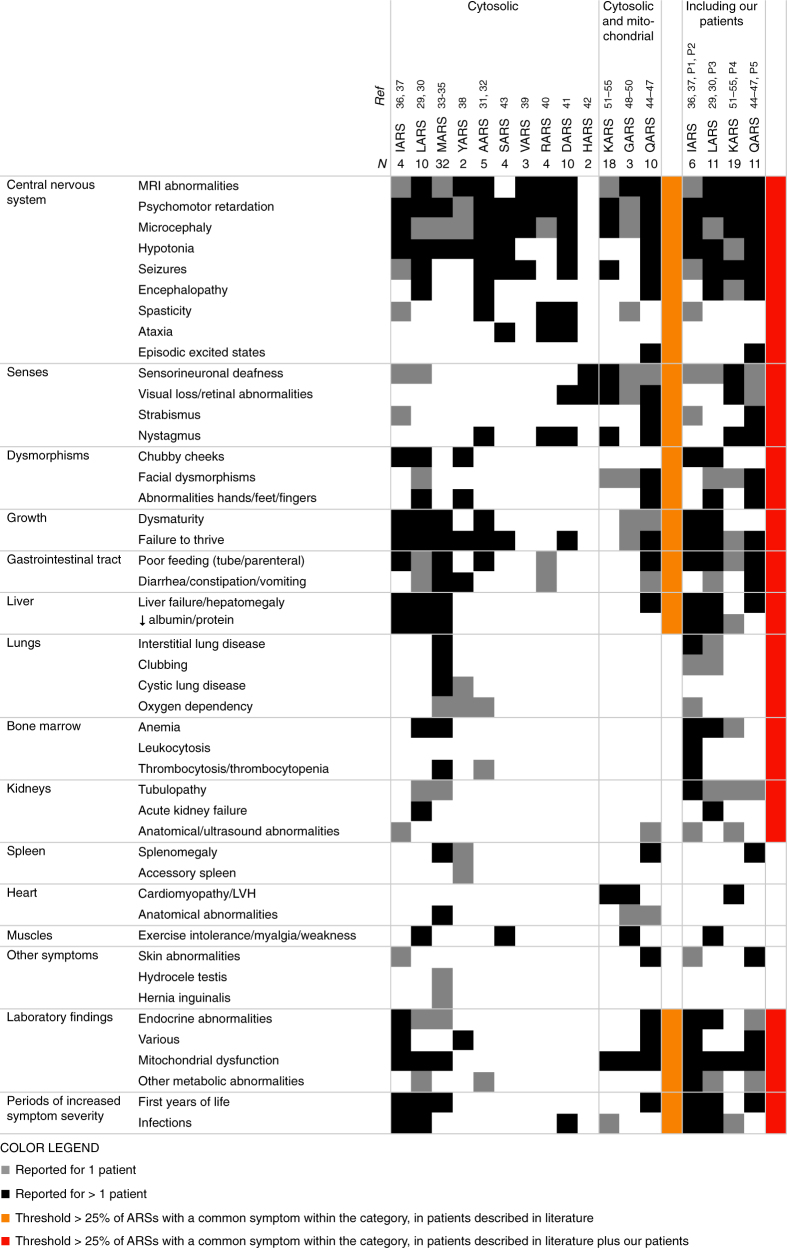
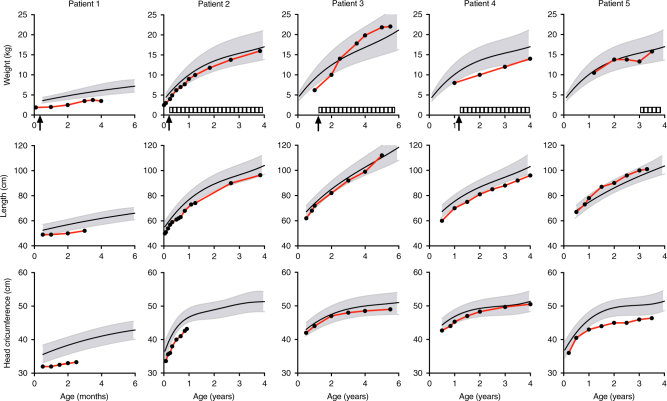
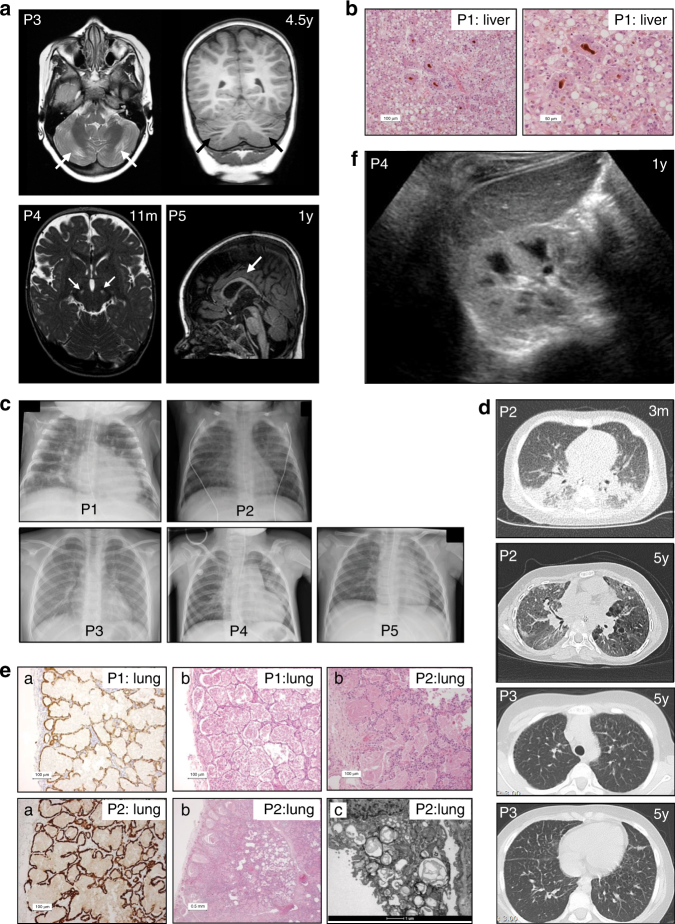
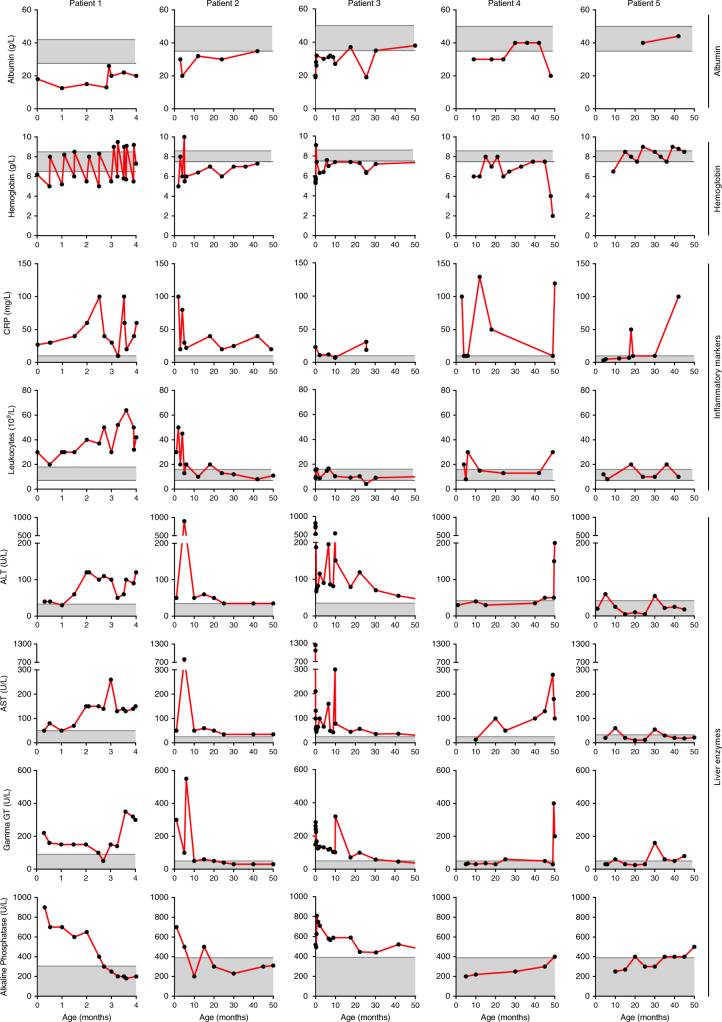
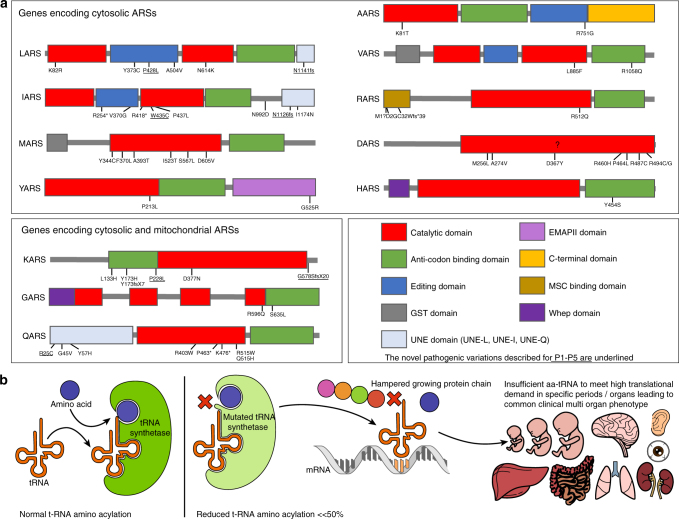
Results: In literature, we identified 107 patients with AARS, DARS, GARS, HARS, IARS, KARS, LARS, MARS, RARS, SARS, VARS, YARS, and QARS deficiencies. Common symptoms (defined as present in ≥4/13 ARS deficiencies) included abnormalities of the central nervous system and/or senses (13/13), failure to thrive, gastrointestinal symptoms, dysmaturity, liver disease, and facial dysmorphisms. Deep phenotyping of 5 additional patients with unreported compound heterozygous pathogenic variations in IARS, LARS, KARS, and QARS extended the common phenotype with lung disease, hypoalbuminemia, anemia, and renal tubulopathy.
Conclusion: We propose a common clinical phenotype for recessive ARS deficiencies, resulting from insufficient aminoacylation activity to meet translational demand in specific organs or periods of life. Assuming residual ARS activity, adequate protein/amino acid supply seems essential instead of the traditional replacement of protein by glucose in patients with metabolic diseases.
Keywords: Aminoacyl-tRNA synthetase deficiency; Clinical phenotype; Cytosolic translation.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
Comment in
-
Correspondence on "Aminoacyl-tRNA synthetase deficiencies in search of common themes" by Fuchs et al.Genet Med. 2021 Mar;23(3):587-588. doi: 10.1038/s41436-020-01013-9. Epub 2020 Oct 22. Genet Med. 2021. PMID: 33087889 No abstract available.
-
Response to Shen and Zou.Genet Med. 2021 Mar;23(3):589-590. doi: 10.1038/s41436-020-01014-8. Epub 2020 Oct 22. Genet Med. 2021. PMID: 33087890 No abstract available.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
