

PRDM16 isoforms differentially regulate normal and leukemic hematopoiesis and inflammatory gene signature
- PMID: 29878897
- PMCID: PMC6063481
- DOI: 10.1172/JCI99862
PRDM16 isoforms differentially regulate normal and leukemic hematopoiesis and inflammatory gene signature
Abstract
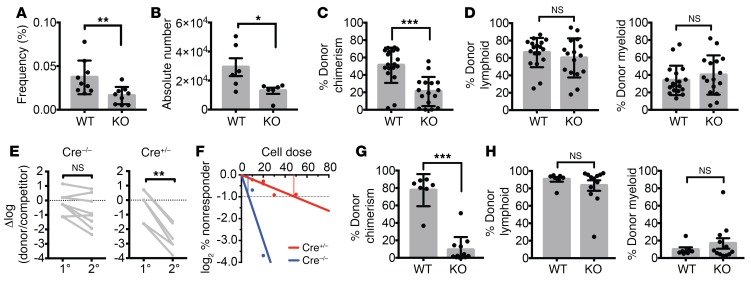
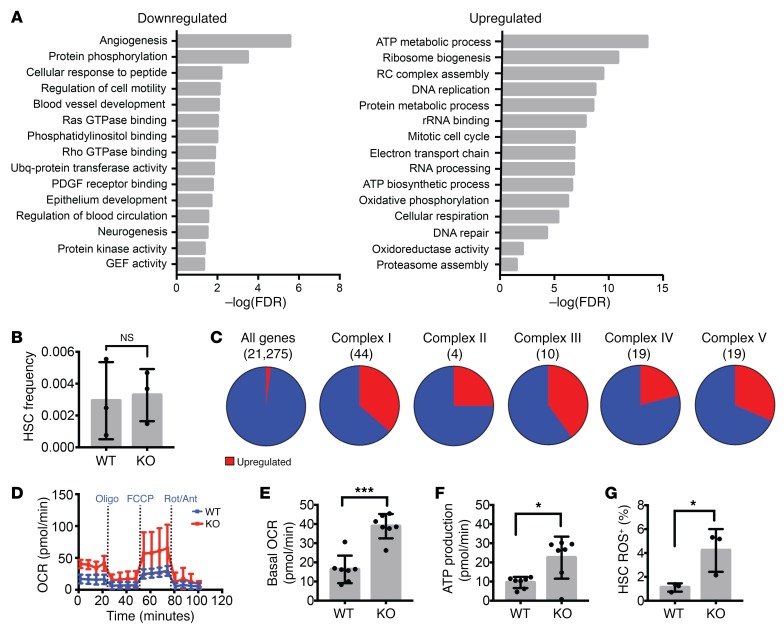
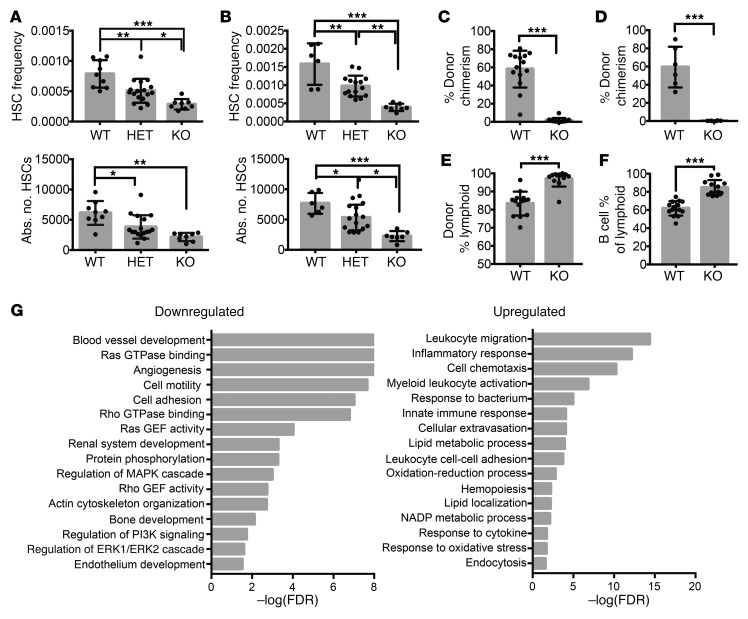
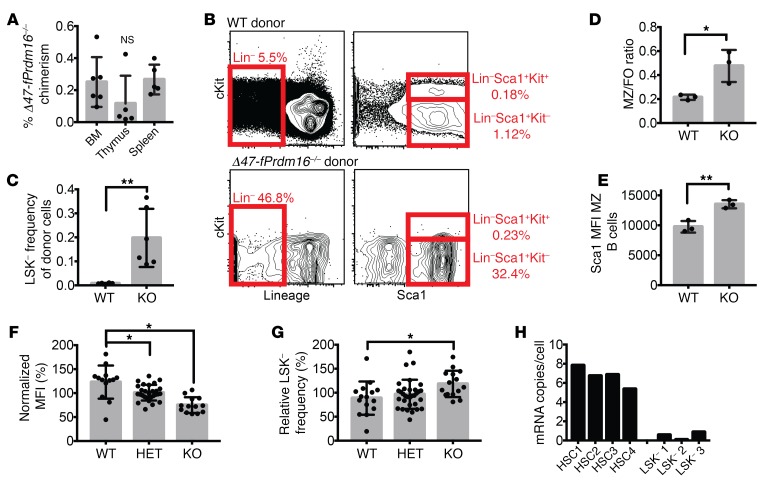
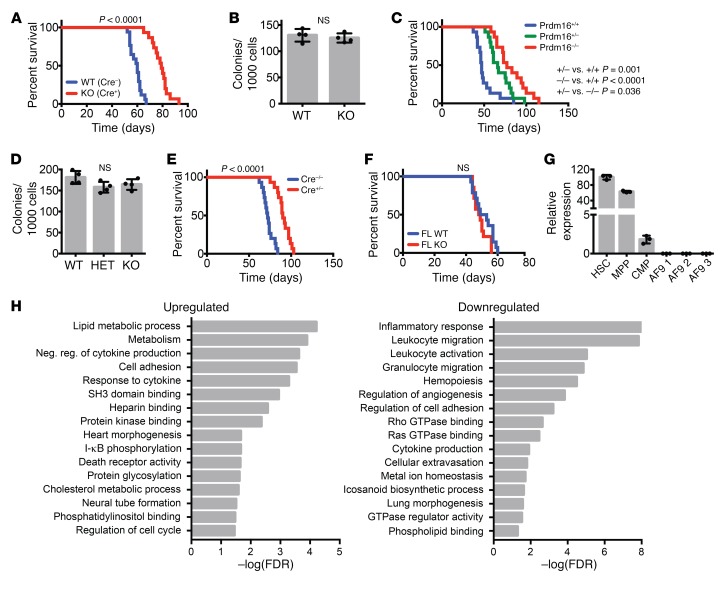
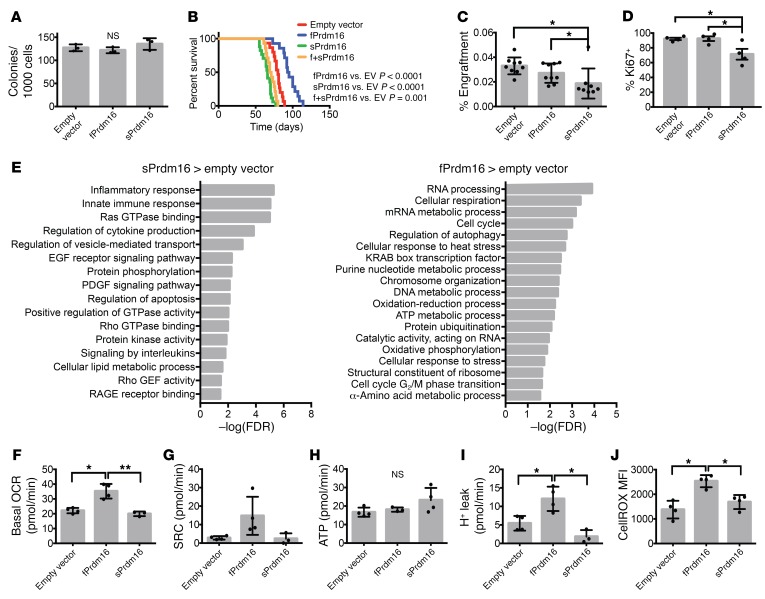
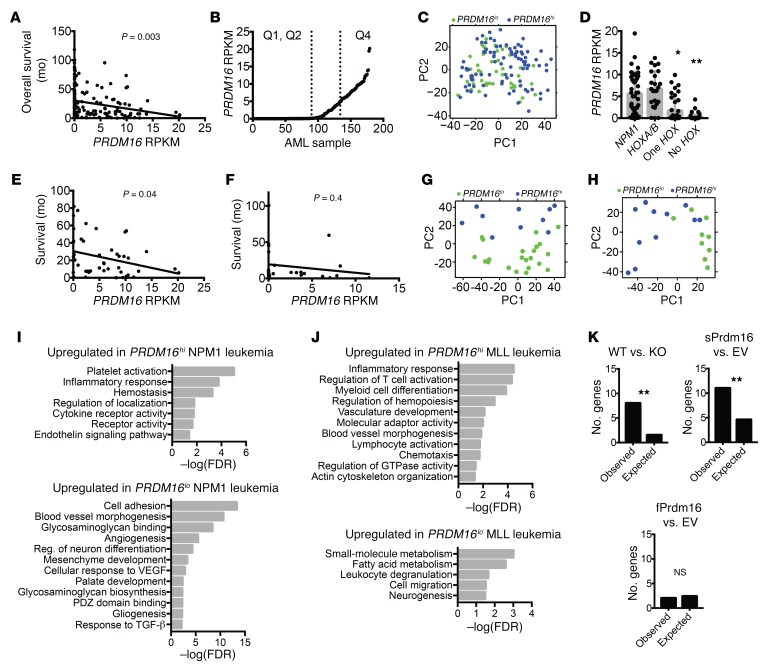
PRDM16 is a transcriptional coregulator involved in translocations in acute myeloblastic leukemia (AML), myelodysplastic syndromes, and T acute lymphoblastic leukemia that is highly expressed in and required for the maintenance of hematopoietic stem cells (HSCs), and can be aberrantly expressed in AML. Prdm16 is expressed as full-length (fPrdm16) and short (sPrdm16) isoforms, the latter lacking the N-terminal PR domain. The role of both isoforms in normal and malignant hematopoiesis is unclear. We show here that fPrdm16 was critical for HSC maintenance, induced multiple genes involved in GTPase signaling, and repressed inflammation, while sPrdm16 supported B cell development biased toward marginal zone B cells and induced an inflammatory signature. In a mouse model of human MLL-AF9 leukemia, fPrdm16 extended latency, while sPrdm16 shortened latency and induced a strong inflammatory signature, including several cytokines and chemokines that are associated with myelodysplasia and with a worse prognosis in human AML. Finally, in human NPM1-mutant and in MLL-translocated AML, high expression of PRDM16, which negatively impacts outcome, was associated with inflammatory gene expression, thus corroborating the mouse data. Our observations demonstrate distinct roles for Prdm16 isoforms in normal HSCs and AML, and identify sPrdm16 as one of the drivers of prognostically adverse inflammation in leukemia.
Keywords: Adult stem cells; Bone marrow; Hematology; Leukemias; Stem cells.
Conflict of interest statement
Figures
References
-
- Mochizuki N, et al. A novel gene, MEL1, mapped to 1p36.3 is highly homologous to the MDS1/EVI1 gene and is transcriptionally activated in t(1;3)(p36;q21)-positive leukemia cells. Blood. 2000;96(9):3209–3214. - PubMed
-
- Moir DJ, Jones PA, Pearson J, Duncan JR, Cook P, Buckle VJ. A new translocation, t(1;3) (p36;q21), in myelodysplastic disorders. Blood. 1984;64(2):553–555. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
