

METHimpute: imputation-guided construction of complete methylomes from WGBS data
- PMID: 29879918
- PMCID: PMC5992726
- DOI: 10.1186/s12864-018-4641-x
METHimpute: imputation-guided construction of complete methylomes from WGBS data
Abstract
Background: Whole-genome bisulfite sequencing (WGBS) has become the standard method for interrogating plant methylomes at base resolution. However, deep WGBS measurements remain cost prohibitive for large, complex genomes and for population-level studies. As a result, most published plant methylomes are sequenced far below saturation, with a large proportion of cytosines having either missing data or insufficient coverage.
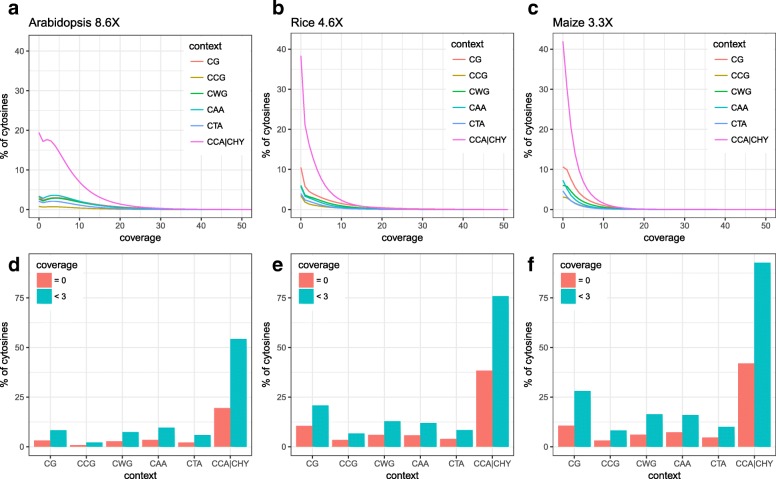
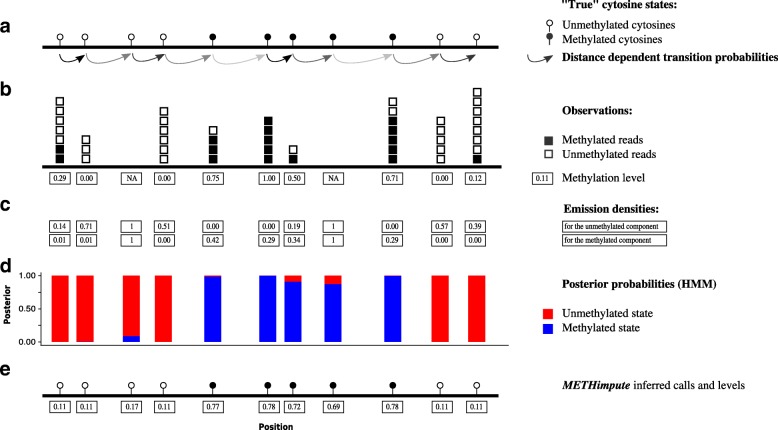
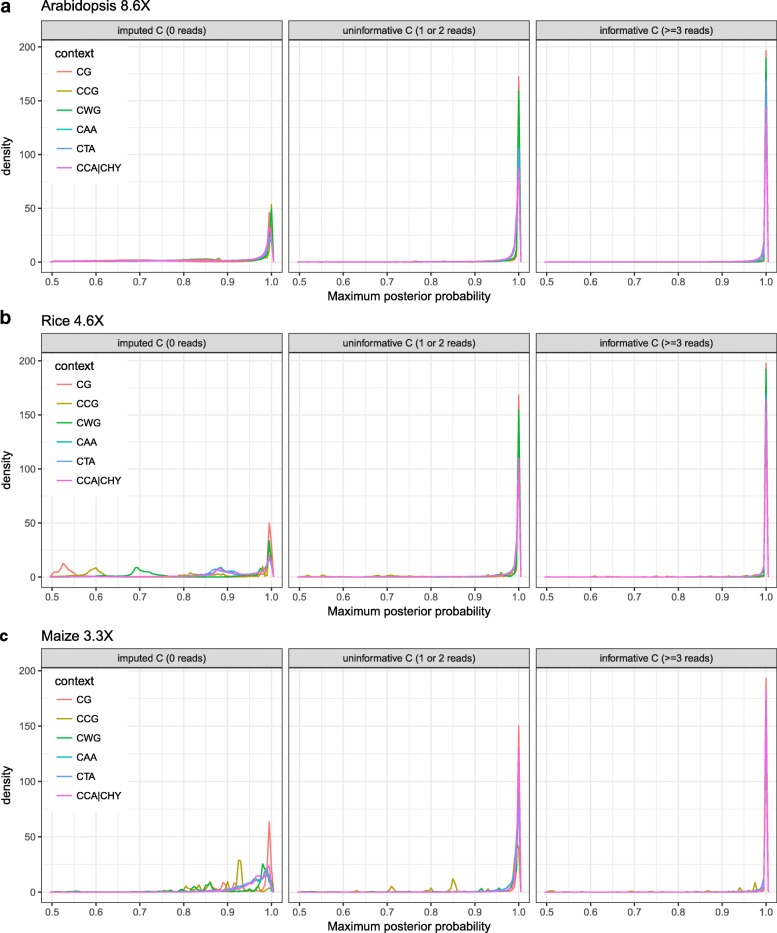
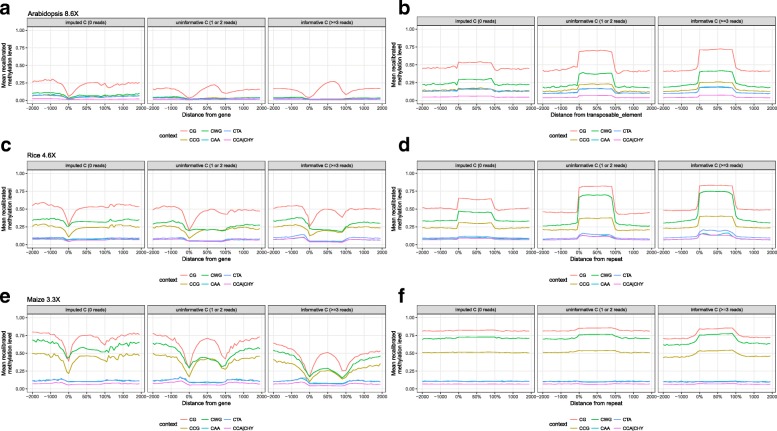
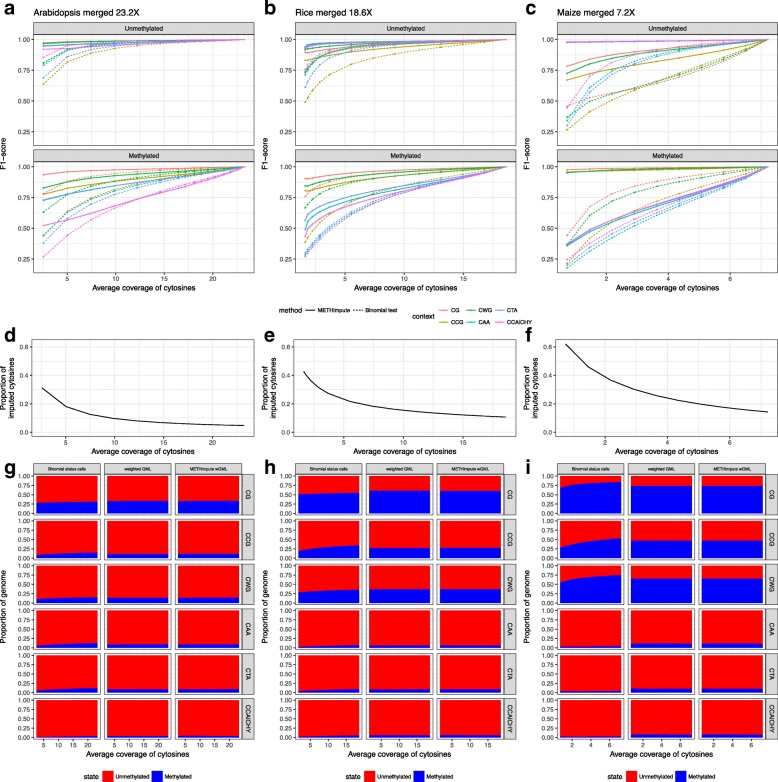
Results: Here we present METHimpute, a Hidden Markov Model (HMM) based imputation algorithm for the analysis of WGBS data. Unlike existing methods, METHimpute enables the construction of complete methylomes by inferring the methylation status and level of all cytosines in the genome regardless of coverage. Application of METHimpute to maize, rice and Arabidopsis shows that the algorithm infers cytosine-resolution methylomes with high accuracy from data as low as 6X, compared to data with 60X, thus making it a cost-effective solution for large-scale studies.
Conclusions: METHimpute provides methylation status calls and levels for all cytosines in the genome regardless of coverage, thus yielding complete methylomes even with low-coverage WGBS datasets. The method has been extensively tested in plants, but should also be applicable to other species. An implementation is available on Bioconductor.
Keywords: Hidden Markov Model; Imputation; Methylation; Whole-genome bisulfite sequencing.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
-
- Feng S, Cokus SJ, Zhang X, Chen P-Y, Bostick M, Goll MG, Hetzel J, Jain J, Strauss SH, Halpern ME, Ukomadu C, Sadler KC, Pradhan S, Pellegrini M, Jacobsen SE. Conservation and divergence of methylation patterning in plants and animals. Proc Natl Acad Sci. 2010;107(19):8689–94. doi: 10.1073/pnas.1002720107. - DOI - PMC - PubMed
-
- Niederhuth CE, Bewick AJ, Ji L, Alabady M, Kim KD, Page JT, Li Q, Rohr NA, Rambani A, Burke JM, Udall JA, Egesi C, Schmutz J, Grimwood J, Jackson SA, Springer NM, Schmitz RJ. Widespread natural variation of DNA methylation within angiosperms. Genome Biol. 2016;17(194). 10.1186/s13059-016-1059-0. - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
