

Accessibility of the Shine-Dalgarno Sequence Dictates N-Terminal Codon Bias in E. coli
- PMID: 29883608
- PMCID: PMC6311106
- DOI: 10.1016/j.molcel.2018.05.008
Accessibility of the Shine-Dalgarno Sequence Dictates N-Terminal Codon Bias in E. coli
Abstract
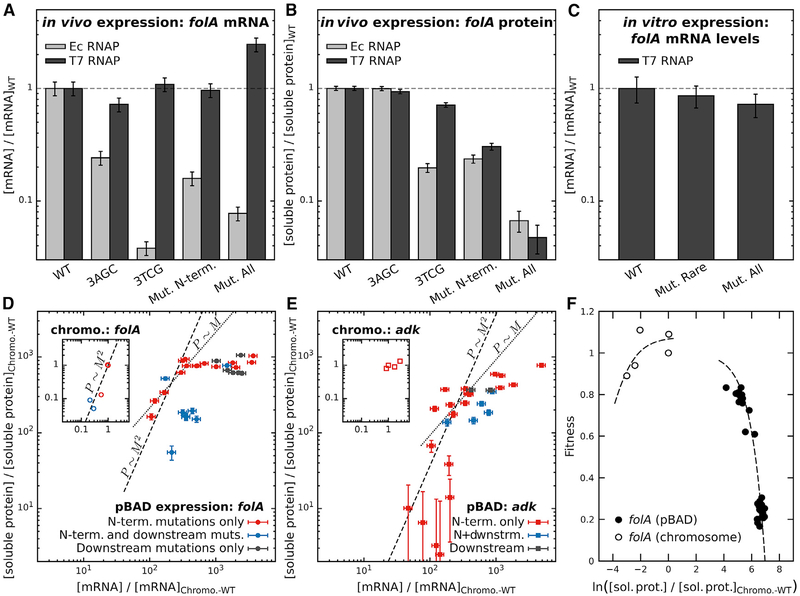
Despite considerable efforts, no physical mechanism has been shown to explain N-terminal codon bias in prokaryotic genomes. Using a systematic study of synonymous substitutions in two endogenous E. coli genes, we show that interactions between the coding region and the upstream Shine-Dalgarno (SD) sequence modulate the efficiency of translation initiation, affecting both intracellular mRNA and protein levels due to the inherent coupling of transcription and translation in E. coli. We further demonstrate that far-downstream mutations can also modulate mRNA levels by occluding the SD sequence through the formation of non-equilibrium secondary structures. By contrast, a non-endogenous RNA polymerase that decouples transcription and translation largely alleviates the effects of synonymous substitutions on mRNA levels. Finally, a complementary statistical analysis of the E. coli genome specifically implicates avoidance of intra-molecular base pairing with the SD sequence. Our results provide general physical insights into the coding-level features that optimize protein expression in prokaryotes.
Copyright © 2018 Elsevier Inc. All rights reserved.
Conflict of interest statement
DECLARATION OF INTERESTS
The authors declare no competing interests.
Figures






Similar articles
-
Importance of the leader region of mRNA for translation initiation of ColE2 Rep protein.Plasmid. 2007 Nov;58(3):249-60. doi: 10.1016/j.plasmid.2007.07.001. Epub 2007 Aug 27. Plasmid. 2007. PMID: 17720244
-
Novel Translation Initiation Regulation Mechanism in Escherichia coli ptrB Mediated by a 5'-Terminal AUG.J Bacteriol. 2017 Jun 27;199(14):e00091-17. doi: 10.1128/JB.00091-17. Print 2017 Jul 15. J Bacteriol. 2017. PMID: 28484048 Free PMC article.
-
The role of bases upstream of the Shine-Dalgarno region and in the coding sequence in the control of gene expression in Escherichia coli: translation and stability of mRNAs in vivo.Gene. 1989 May 15;78(1):59-72. doi: 10.1016/0378-1119(89)90314-4. Gene. 1989. PMID: 2475391
-
Codon influence on protein expression in E. coli correlates with mRNA levels.Nature. 2016 Jan 21;529(7586):358-363. doi: 10.1038/nature16509. Epub 2016 Jan 13. Nature. 2016. PMID: 26760206 Free PMC article.
-
The diversity of Shine-Dalgarno sequences sheds light on the evolution of translation initiation.RNA Biol. 2021 Nov;18(11):1489-1500. doi: 10.1080/15476286.2020.1861406. Epub 2020 Dec 21. RNA Biol. 2021. PMID: 33349119 Free PMC article. Review.
Cited by
-
A genetic selection reveals functional metastable structures embedded in a toxin-encoding mRNA.Elife. 2019 Aug 14;8:e47549. doi: 10.7554/eLife.47549. Elife. 2019. PMID: 31411564 Free PMC article.
-
Reprogramming the genetic code.Nat Rev Genet. 2021 Mar;22(3):169-184. doi: 10.1038/s41576-020-00307-7. Epub 2020 Dec 14. Nat Rev Genet. 2021. PMID: 33318706 Review.
-
Genes for highly abundant proteins in Escherichia coli avoid 5' codons that promote ribosomal initiation.PLoS Comput Biol. 2023 Oct 25;19(10):e1011581. doi: 10.1371/journal.pcbi.1011581. eCollection 2023 Oct. PLoS Comput Biol. 2023. PMID: 37878567 Free PMC article.
-
Comprehensive exploration of the translocation, stability and substrate recognition requirements in VIM-2 lactamase.Elife. 2020 Jun 8;9:e56707. doi: 10.7554/eLife.56707. Elife. 2020. PMID: 32510322 Free PMC article.
-
The sRNA DicF integrates oxygen sensing to enhance enterohemorrhagic Escherichia coli virulence via distinctive RNA control mechanisms.Proc Natl Acad Sci U S A. 2019 Jul 9;116(28):14210-14215. doi: 10.1073/pnas.1902725116. Epub 2019 Jun 24. Proc Natl Acad Sci U S A. 2019. PMID: 31235565 Free PMC article.
References
-
- Adhya S, and Gottesman M (1978). Control of transcription termination. Annu. Rev. Biochem 47, 967–996. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
