

HSP60 critically regulates endogenous IL-1β production in activated microglia by stimulating NLRP3 inflammasome pathway
- PMID: 29885667
- PMCID: PMC5994257
- DOI: 10.1186/s12974-018-1214-5
HSP60 critically regulates endogenous IL-1β production in activated microglia by stimulating NLRP3 inflammasome pathway
Erratum in
-
Correction to: HSP60 critically regulates endogenous IL-1β production in activated microglia by stimulating NLRP3 inflammasome pathway.J Neuroinflammation. 2018 Nov 15;15(1):317. doi: 10.1186/s12974-018-1355-6. J Neuroinflammation. 2018. PMID: 30442172 Free PMC article.
Abstract
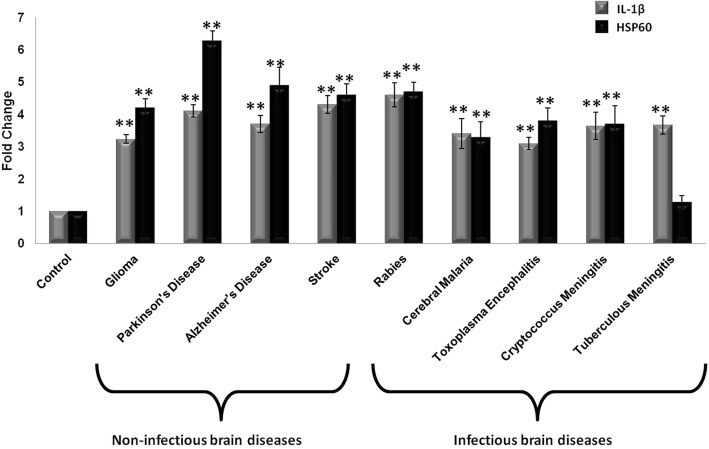
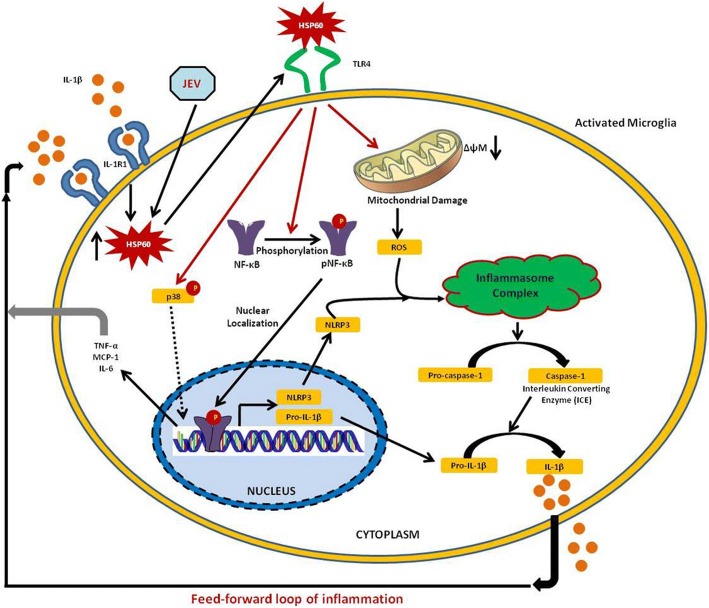
Background: Interleukin-1β (IL-1β) is one of the most important cytokine secreted by activated microglia as it orchestrates the vicious cycle of inflammation by inducing the expression of various other pro-inflammatory cytokines along with its own production. Microglia-mediated IL-1β production is a tightly regulated mechanism which involves the activation of nucleotide-binding oligomerization domain leucine-rich repeat and pyrin domain-containing 3 (NLRP3) inflammasome pathway. Our previous study suggests the critical role of heat shock protein 60 (HSP60) in IL-1β-induced inflammation in microglia through TLR4-p38 MAPK axis. However, whether HSP60 regulates endogenous IL-1β production is not known. Therefore, to probe the underlying mechanism, we elucidate the role of HSP60 in endogenous IL-1β production.
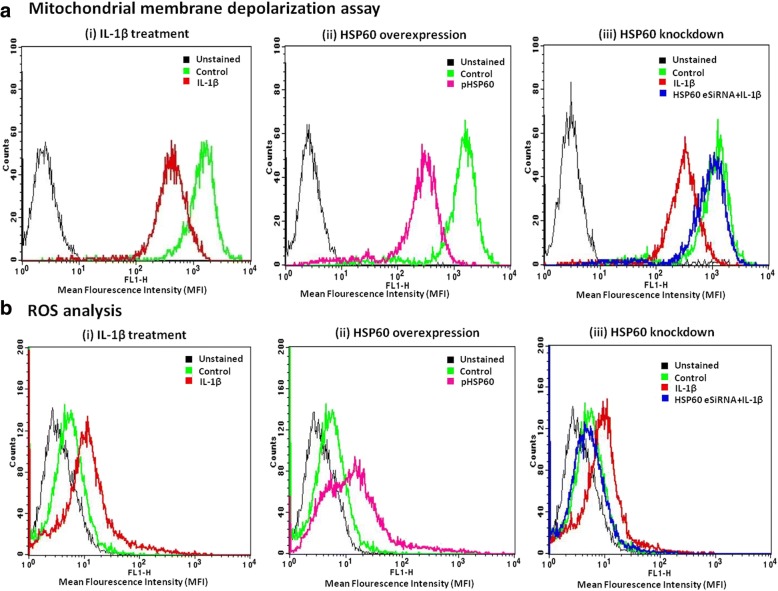
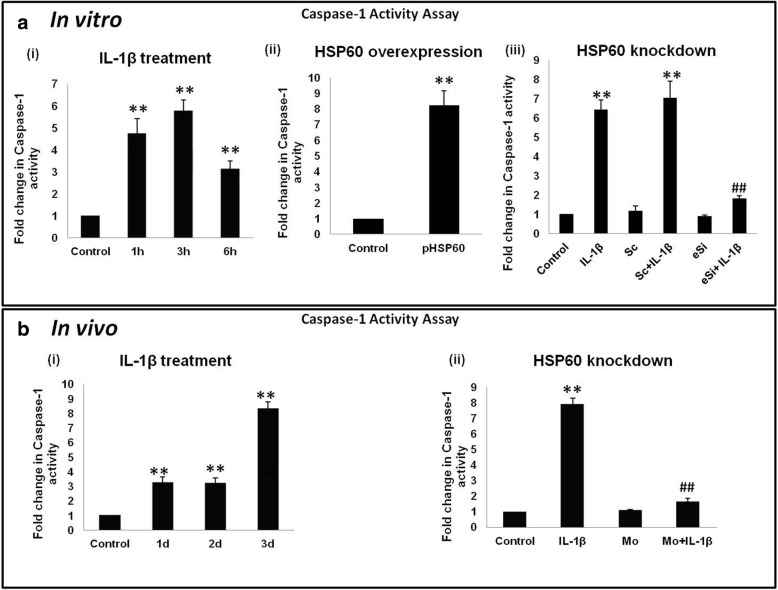
Methods: We used in vitro (N9 murine microglial cells) and in vivo (BALB/c mouse) models for our study. HSP60 overexpression and knockdown experiment was done to elucidate the role of HSP60 in endogenous IL-1β production by microglia. Western blotting and quantitative real-time PCR was performed using N9 cells and BALB/c mice brain, to analyze various proteins and transcript levels. Reactive oxygen species levels and mitochondrial membrane depolarization in N9 cells were analyzed by flow cytometry. We also performed caspase-1 activity assay and enzyme-linked immunosorbent assay to assess caspase-1 activity and IL-1β production, respectively.
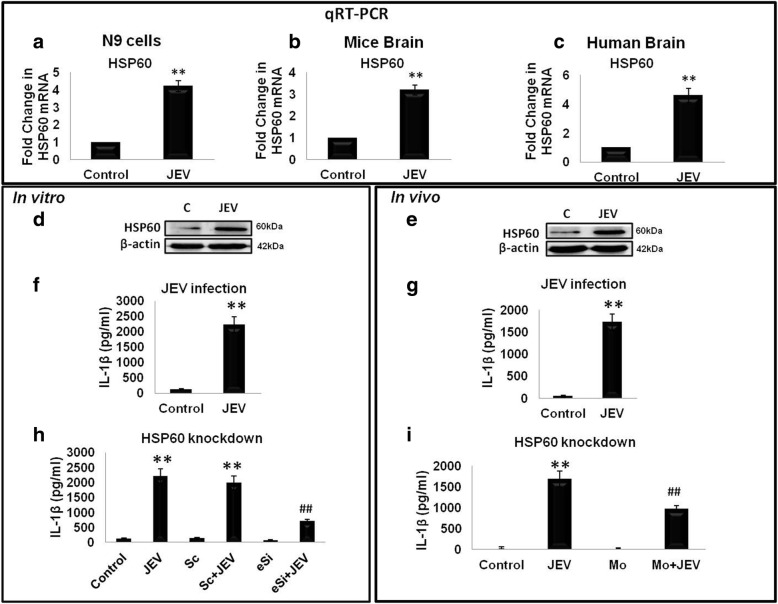
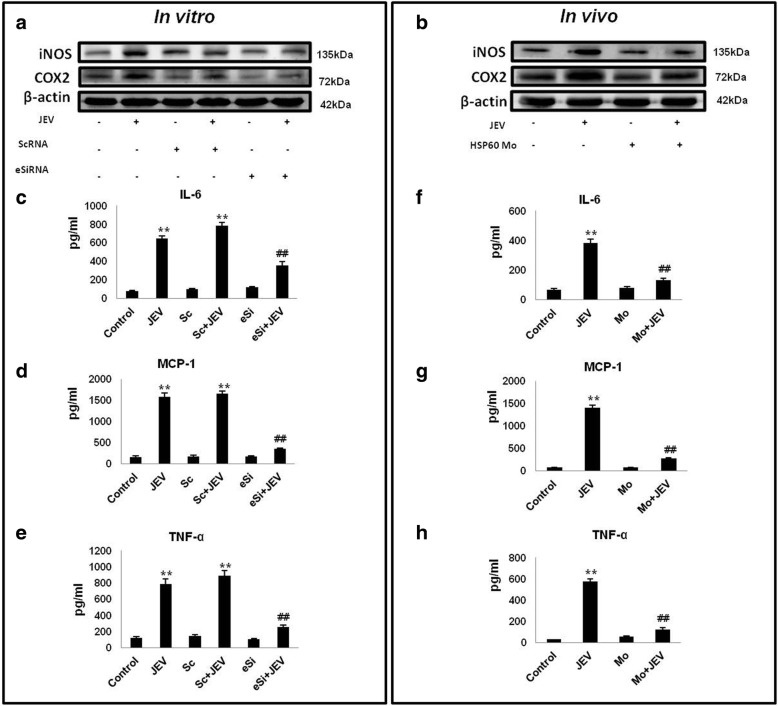
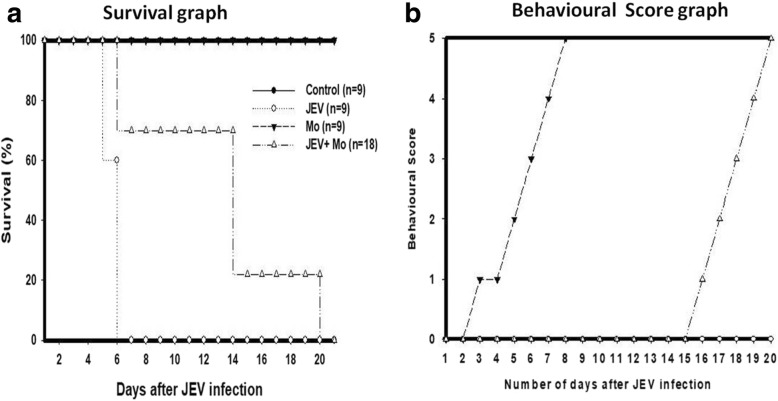
Results: HSP60 induces the phosphorylation and nuclear localization of NF-κB both in vitro and in vivo. It also induces perturbation in mitochondrial membrane potential and enhances reactive oxygen species (ROS) generation in microglia. HSP60 further activates NLRP3 inflammasome by elevating NLRP3 expression both at RNA and protein levels. Furthermore, HSP60 enhances caspase-1 activity and increases IL-1β secretion by microglia. Knockdown of HSP60 reduces the IL-1β-induced production of IL-1β both in vitro and in vivo. Also, we have shown for the first time that knockdown of HSP60 leads to decreased IL-1β production during Japanese encephalitis virus (JEV) infection, which eventually leads to decreased inflammation and increased survival of JEV-infected mice.
Conclusion: HSP60 mediates microglial IL-1β production by regulating NLRP3 inflammasome pathway and reduction of HSP60 leads to reduction of inflammation in JEV infection.
Keywords: Caspase-1; HSP60; Heat shock protein; IL-1β; Inflammasome; Inflammation; Japanese encephalitis virus (JEV); Microglia; Mitochondrial stress; NLRP3; ROS.
Conflict of interest statement
Ethics approval
Institutional Animal Ethics Committee of the National Brain Research Centre (NBRC) approved the protocol of the study (NBRC/IAEC/2016/115 and NBRC/IAEC/2017/028).
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures




References
-
- Lehnardt S. Innate immunity and neuroinflammation in the CNS: the role of microglia in toll-like receptor-mediated neuronal injury. Glia. 2010;58:253–263. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
