

Cancer: From Wild-Type to Mutant Huntingtin
- PMID: 29889077
- PMCID: PMC6087435
- DOI: 10.3233/JHD-180290
Cancer: From Wild-Type to Mutant Huntingtin
Abstract
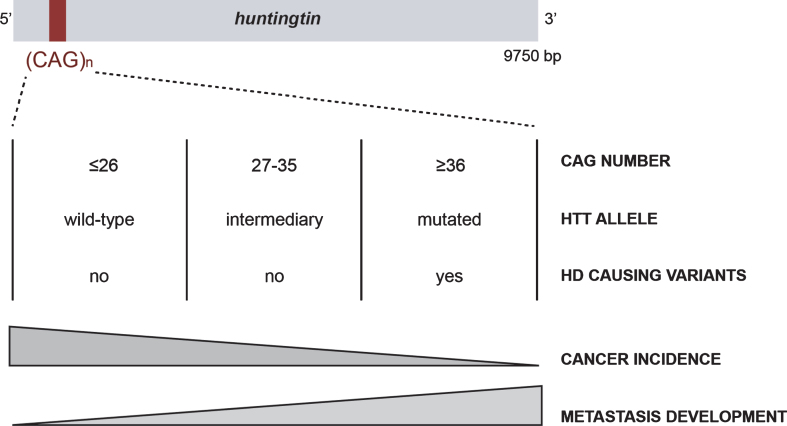
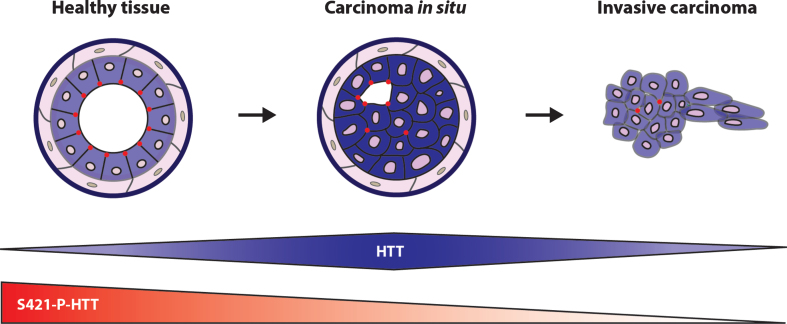
Huntingtin (HTT) is a scaffold protein mostly known because it gives rise to the severe and incurable inherited neurological disorder Huntington's disease (HD) when mutated. The Huntingtin gene (HTT) carries a polymorphic trinucleotide expansion of CAGs in exon 1 that ranges from 9 to 35 in the non-HD affected population. However, if it exceeds 35 CAG repeats, the altered protein is referred to as mutant HTT and leads to the development of HD. Given the wide spectrum of severe symptoms developed by HD individuals, wild-type and mutant HTT have been mostly studied in the context of this disorder. However, HTT expression is ubiquitous and several peripheral symptoms in HD have been described, suggesting that HTT is of importance, not only in the central nervous system (CNS), but also in peripheral organs. Accordingly, HTT and mutant HTT may interfere with non-brain-related diseases. Correlative studies have highlighted a decreased cancer incidence in the HD population and both wild-type and mutant HTT have been implicated in tumor progression. In this review, we describe the current evidence linking wild-type and mutant HTT to cancer and discuss how CAG polymorphism, HTT function, and partners may influence carcinogenesis and metastatic progression.
Keywords: CAG expansion; Huntington disease; adhesion; breast cancer; cancer; huntingtin; metastasis.
Figures
References
-
- The Huntington’s Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. 1993;72(6):971–83. - PubMed
-
- Kremer B, Goldberg P, Andrew SE, Theilmann J, Telenius H, Zeisler J, et al. A worldwide study of the Huntington’s disease mutation, The sensitivity and specificity of measuring CAG repeats. N Engl J Med. 1994;330(20):1401–6. - PubMed
-
- Rubinsztein DC, Leggo J, Coles R, Almqvist E, Biancalana V, Cassiman JJ, et al. Phenotypic characterization of individuals with 30-40 CAG repeats in the Huntington disease (HD) gene reveals HD cases with 36 repeats and apparently normal elderly individuals with 36-39 repeats. Am J Hum Genet, 1996;59(1):16–22. - PMC - PubMed
-
- Andrew SE, Goldberg YP, Kremer B, Telenius H, Theilmann J, Adam S, et al. The relationship between trinucleotide (CAG) repeat length and clinical features of Huntington’s disease. Nat Genet, 1993;4(4):398–403. - PubMed
-
- Snell RG, MacMillan JC, Cheadle JP, Fenton I, Lazarou LP, Davies P, et al. Relationship between trinucleotide repeat expansion and phenotypic variation in Huntington’s disease. Nat Genet, 1993;4(4):393–7. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
