

The Roles of p53 in Mitochondrial Dynamics and Cancer Metabolism: The Pendulum between Survival and Death in Breast Cancer?
- PMID: 29890631
- PMCID: PMC6024909
- DOI: 10.3390/cancers10060189
The Roles of p53 in Mitochondrial Dynamics and Cancer Metabolism: The Pendulum between Survival and Death in Breast Cancer?
Abstract
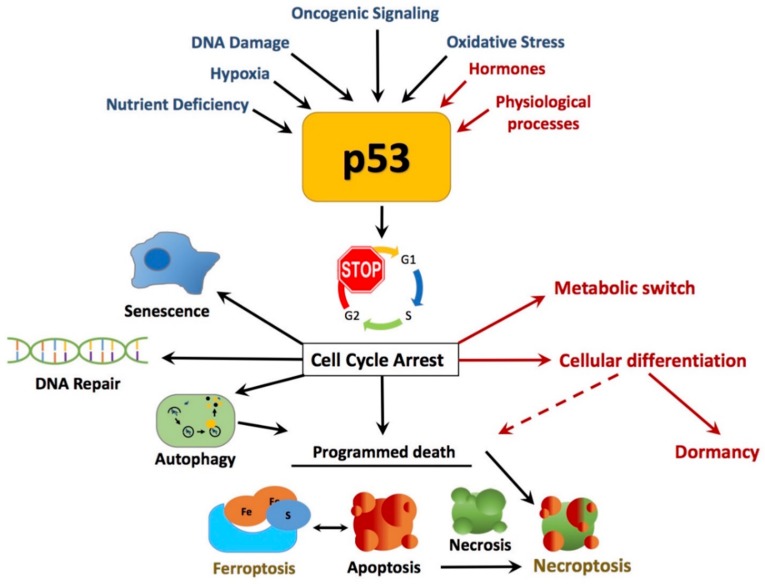
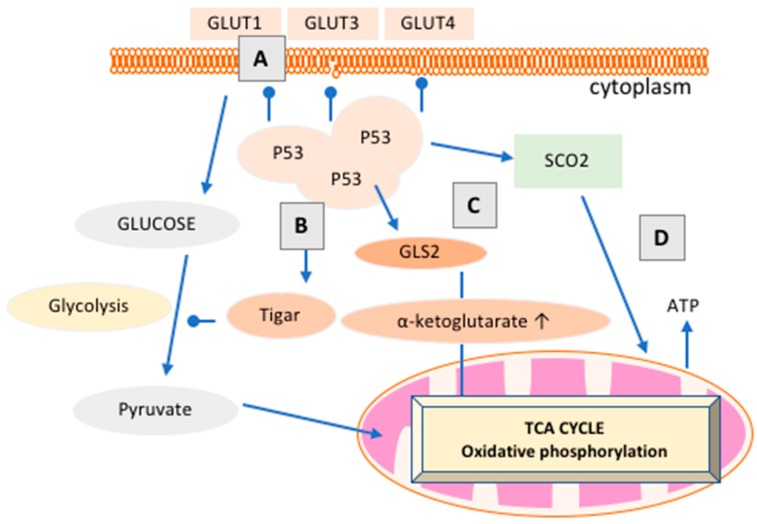
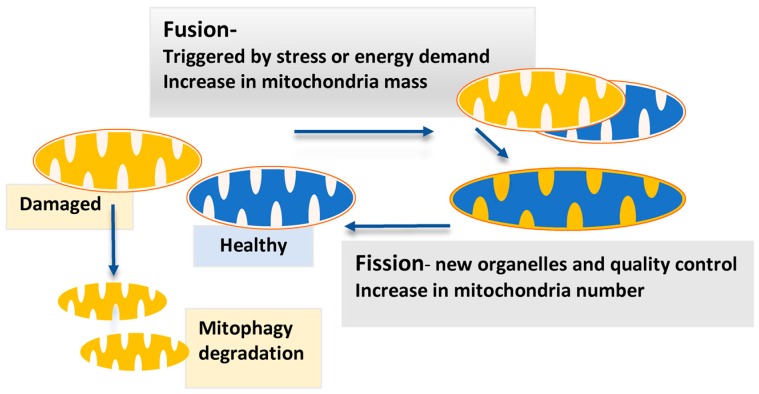
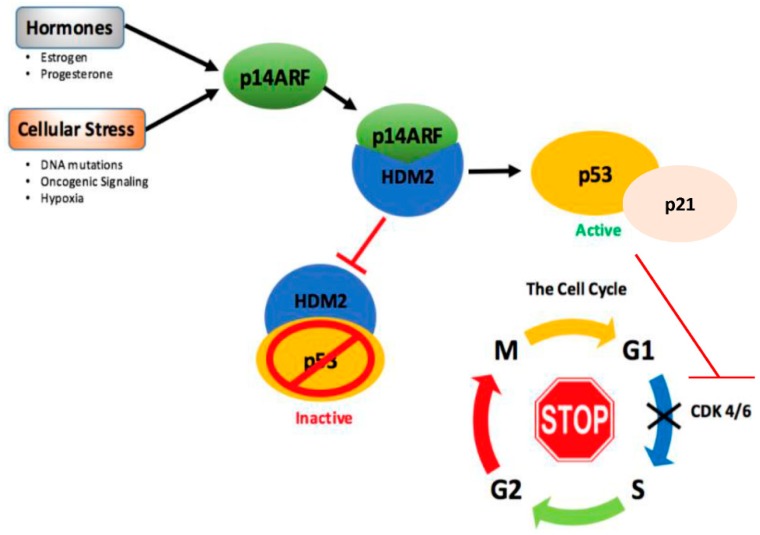
Cancer research has been heavily geared towards genomic events in the development and progression of cancer. In contrast, metabolic regulation, such as aberrant metabolism in cancer, is poorly understood. Alteration in cellular metabolism was once regarded simply as a consequence of cancer rather than as playing a primary role in cancer promotion and maintenance. Resurgence of cancer metabolism research has identified critical metabolic reprogramming events within biosynthetic and bioenergetic pathways needed to fulfill the requirements of cancer cell growth and maintenance. The tumor suppressor protein p53 is emerging as a key regulator of metabolic processes and metabolic reprogramming in cancer cells—balancing the pendulum between cell death and survival. This review provides an overview of the classical and emerging non-classical tumor suppressor roles of p53 in regulating mitochondrial dynamics: mitochondrial engagement in cell death processes in the prevention of cancer. On the other hand, we discuss p53 as a key metabolic switch in cellular function and survival. The focus is then on the conceivable roles of p53 in breast cancer metabolism. Understanding the metabolic functions of p53 within breast cancer metabolism will, in due course, reveal critical metabolic hotspots that cancers advantageously re-engineer for sustenance. Illustration of these events will pave the way for finding novel therapeutics that target cancer metabolism and serve to overcome the breast cancer burden.
Keywords: breast cancer; metabolism; mitochondria; p14ARF; p53; tumor suppressor protein.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
