

Variant site strain typer (VaST): efficient strain typing using a minimal number of variant genomic sites
- PMID: 29890941
- PMCID: PMC5996513
- DOI: 10.1186/s12859-018-2225-z
Variant site strain typer (VaST): efficient strain typing using a minimal number of variant genomic sites
Abstract
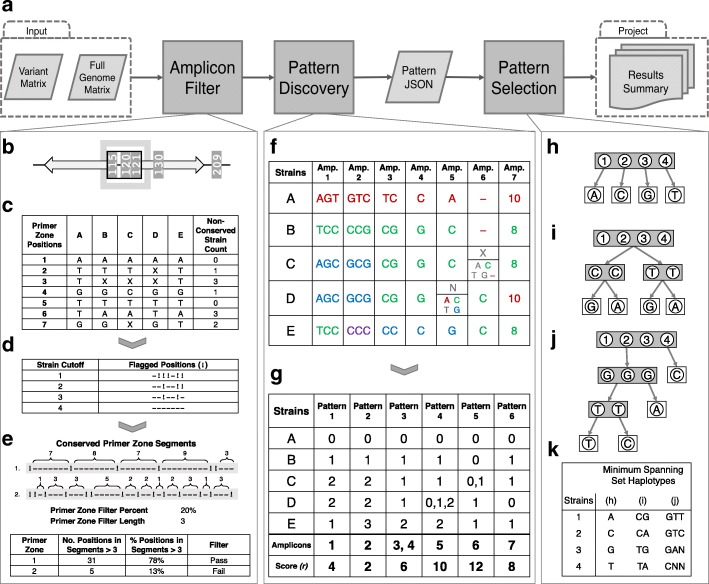
Background: Targeted PCR amplicon sequencing (TAS) techniques provide a sensitive, scalable, and cost-effective way to query and identify closely related bacterial species and strains. Typically, this is accomplished by targeting housekeeping genes that provide resolution down to the family, genera, and sometimes species level. Unfortunately, this level of resolution is not sufficient in many applications where strain-level identification of bacteria is required (biodefense, forensics, clinical diagnostics, and outbreak investigations). Adding more genomic targets will increase the resolution, but the challenge is identifying the appropriate targets. VaST was developed to address this challenge by finding the minimum number of targets that, in combination, achieve maximum strain-level resolution for any strain complex. The final combination of target regions identified by the algorithm produce a unique haplotype for each strain which can be used as a fingerprint for identifying unknown samples in a TAS assay. VaST ensures that the targets have conserved primer regions so that the targets can be amplified in all of the known strains and it also favors the inclusion of targets with basal variants which makes the set more robust when identifying previously unseen strains.
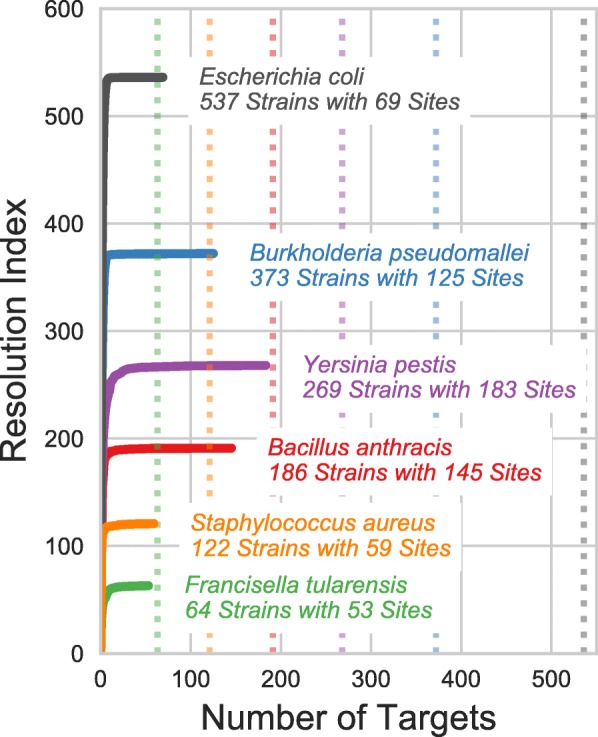
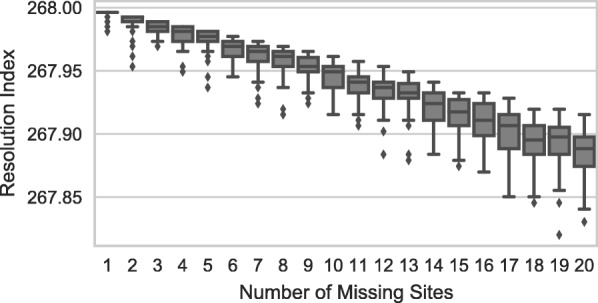
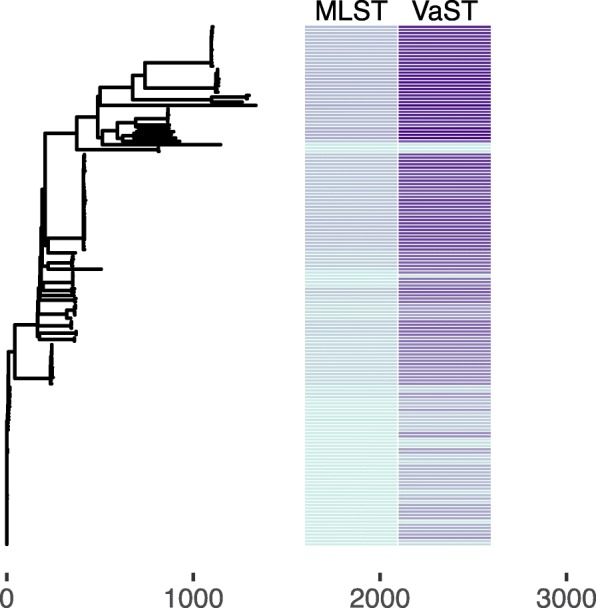
Results: We analyzed VaST's performance using a number of different pathogenic species that are relevant to human disease outbreaks and biodefense. The number of targets required to achieve full resolution ranged from 20 to 88% fewer sites than what would be required in the worst case and most of the resolution is achieved within the first 20 targets. We computationally and experimentally validated one of the VaST panels and found that the targets led to accurate phylogenetic placement of strains, even when the strains were not a part of the original panel design.
Conclusions: VaST is an open source software that, when provided a set of variant sites, can find the minimum number of sites that will provide maximum resolution of a strain complex, and it has many different run-time options that can accommodate a wide range of applications. VaST can be an effective tool in the design of strain identification panels that, when combined with TAS technologies, offer an efficient and inexpensive strain typing protocol.
Keywords: Bacterial strain typing; Single nucleotide polymorphisms; Targeted PCR Amplicon sequencing.
Conflict of interest statement
Ethics approval and consent to participate
Not applicable.
Competing interests
TNF, JWS, and VYF declare that they have applied for a patent for the truncated
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
References
-
- Brzuszkiewicz E, Thürmer A, Schuldes J, Leimbach A, Liesegang H, Meyer F, et al. Genome sequence analyses of two isolates from the recent Escherichia coli outbreak in Germany reveal the emergence of a new pathotype: Entero-Aggregative-Haemorrhagic Escherichia coli (EAHEC). Arch Microbiol. 2011; 193(12):883–91. Available from: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3219860/. - PMC - PubMed
-
- Deng X, den Bakker HC, Hendriksen RS. Genomic Epidemiology: Whole-Genome-Sequencing Powered Surveillance and Outbreak Investigation of Foodborne Bacterial Pathogens. Annu Rev Food Sci Technol. 2016; 7(1):353–74. PMID: 26772415 Available from: 10.1146/annurev-food-041715-033259. - PubMed
-
- Pires dos Santos T, Damborg P, Moodley A, Guardabassi L. Systematic Review on Global Epidemiology of Methicillin-Resistant Staphylococcus pseudintermedius: Inference of Population Structure from Multilocus Sequence Typing Data. Front Microbiol. 2016; 7:1599. Available from: https://www.frontiersin.org/article/10.3389/fmicb.2016.01599. - DOI - PMC - PubMed
-
- Schmedes SE, Sajantila A, Budowle B. Expansion of Microbial Forensics. J Clin Microbiol. 2016; 54(8):1964–74. Available from: http://jcm.asm.org/content/54/8/1964.abstract. - PMC - PubMed
Publication types
MeSH terms
Associated data
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
