

Suppression of detyrosinated microtubules improves cardiomyocyte function in human heart failure
- PMID: 29892068
- PMCID: PMC6195768
- DOI: 10.1038/s41591-018-0046-2
Suppression of detyrosinated microtubules improves cardiomyocyte function in human heart failure
Abstract
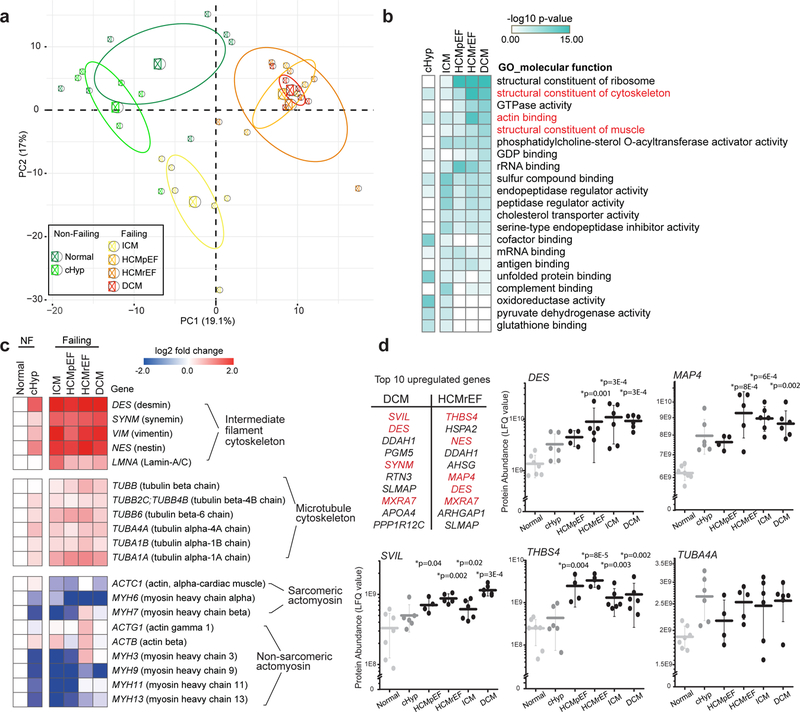
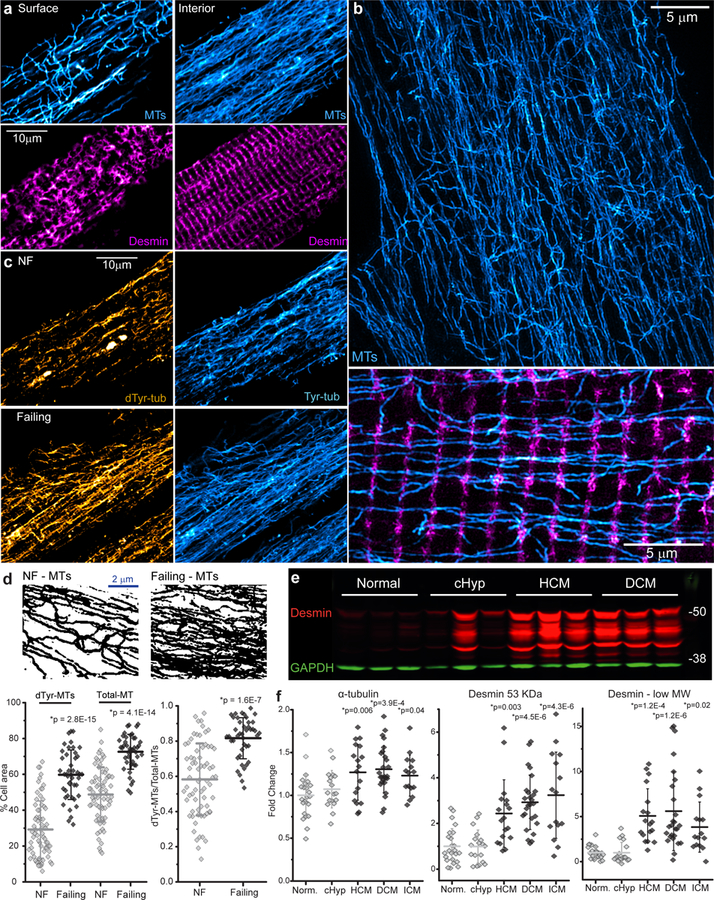
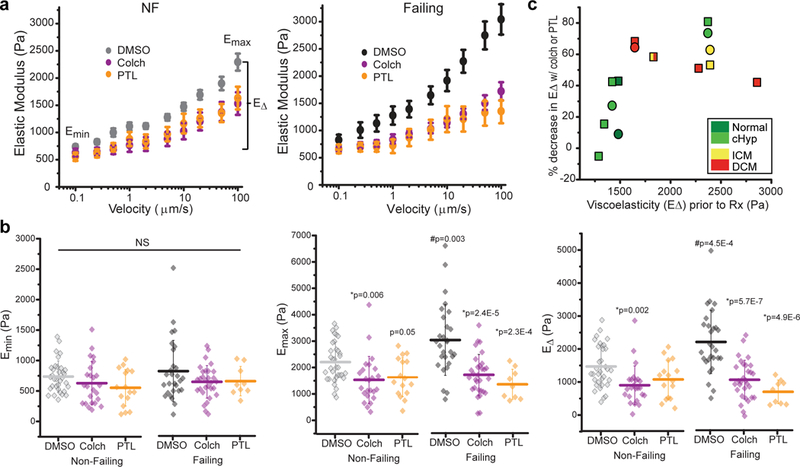
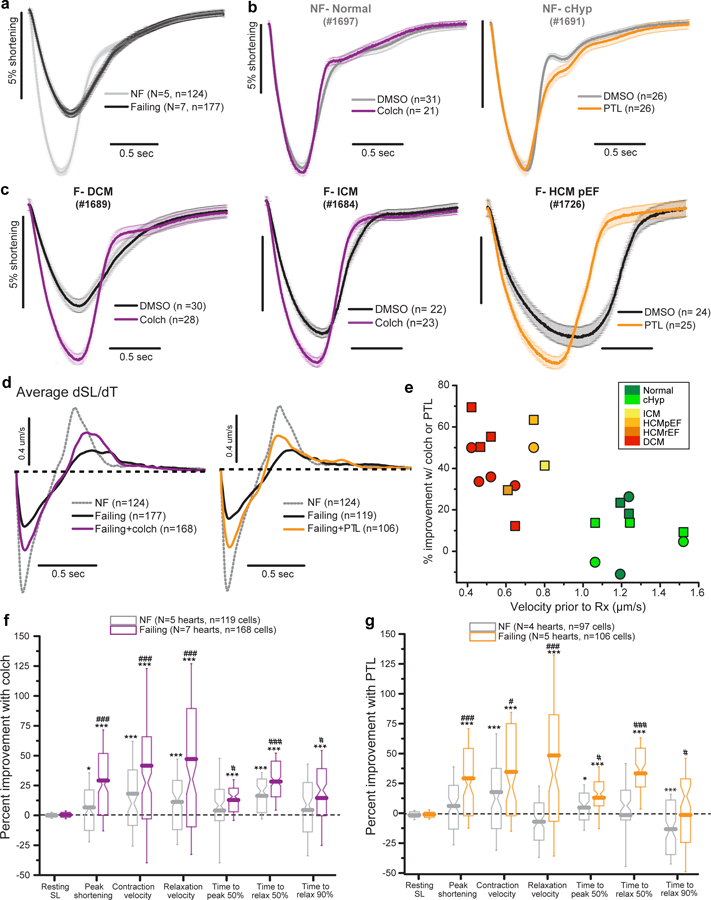
Detyrosinated microtubules provide mechanical resistance that can impede the motion of contracting cardiomyocytes. However, the functional effects of microtubule detyrosination in heart failure or in human hearts have not previously been studied. Here, we utilize mass spectrometry and single-myocyte mechanical assays to characterize changes to the cardiomyocyte cytoskeleton and their functional consequences in human heart failure. Proteomic analysis of left ventricle tissue reveals a consistent upregulation and stabilization of intermediate filaments and microtubules in failing human hearts. As revealed by super-resolution imaging, failing cardiomyocytes are characterized by a dense, heavily detyrosinated microtubule network, which is associated with increased myocyte stiffness and impaired contractility. Pharmacological suppression of detyrosinated microtubules lowers the viscoelasticity of failing myocytes and restores 40-50% of lost contractile function; reduction of microtubule detyrosination using a genetic approach also softens cardiomyocytes and improves contractile kinetics. Together, these data demonstrate that a modified cytoskeletal network impedes contractile function in cardiomyocytes from failing human hearts and that targeting detyrosinated microtubules could represent a new inotropic strategy for improving cardiac function.
Conflict of interest statement
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
Figures
Comment in
-
Targeting the cytoskeleton in heart failure.Nat Rev Cardiol. 2018 Sep;15(9):503. doi: 10.1038/s41569-018-0056-2. Nat Rev Cardiol. 2018. PMID: 29955174 No abstract available.
References
-
- Tsutsui H, Ishihara K & Cooper G Cytoskeletal role in the contractile dysfunction of hypertrophied myocardium. Science 260, 682–687 (1993). - PubMed
-
- Collins JF et al. The role of the cytoskeleton in left ventricular pressure overload hypertrophy and failure. J. Mol. Cell. Cardiol 28, 1435–1443 (1996). - PubMed
-
- Tagawa H et al. Cytoskeletal role in the transition from compensated to decompensated hypertrophy during adult canine left ventricular pressure overloading. Cir. Res 82, 751–761 (1998). - PubMed
-
- Ishibashi Y et al. Role of microtubules in myocyte contractile dysfunction during cardiac hypertrophy in the rat. Am. J. Physiol 271, H1978–87 (1996). - PubMed
-
- Bailey BA, Dipla K, Li S & Houser SR Cellular basis of contractile derangements of hypertrophied feline ventricular myocytes. J. Mol. Cell. Cardiol 29, 1823–1835 (1997). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
