

Abnormalities in Skeletal Muscle Myogenesis, Growth, and Regeneration in Myotonic Dystrophy
- PMID: 29892259
- PMCID: PMC5985300
- DOI: 10.3389/fneur.2018.00368
Abnormalities in Skeletal Muscle Myogenesis, Growth, and Regeneration in Myotonic Dystrophy
Abstract
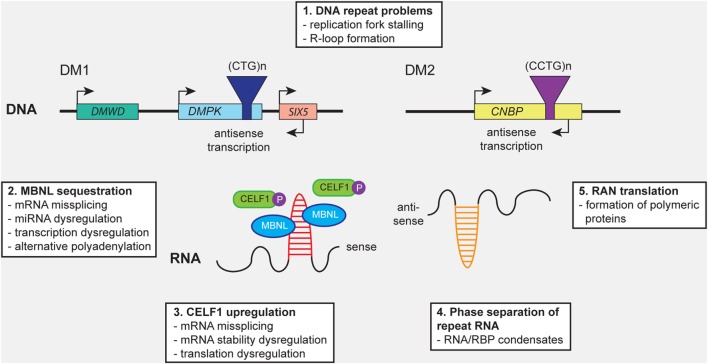
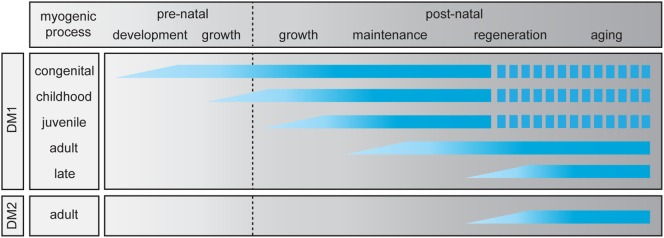
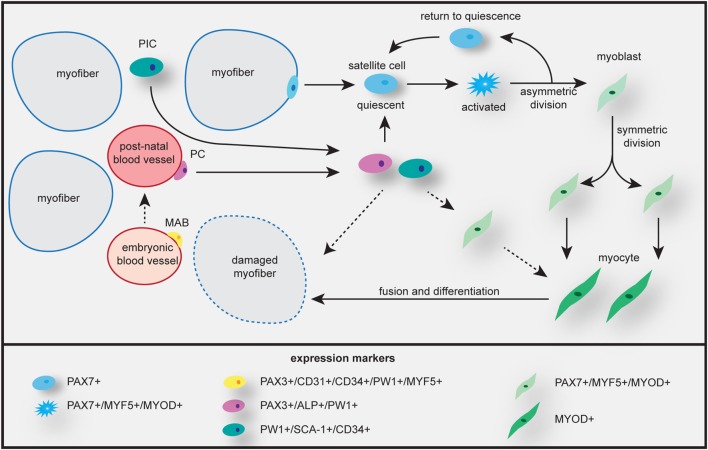
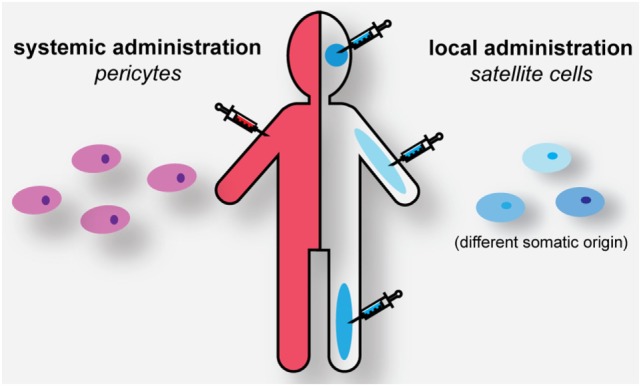
Myotonic dystrophy type 1 (DM1) and 2 (DM2) are autosomal dominant degenerative neuromuscular disorders characterized by progressive skeletal muscle weakness, atrophy, and myotonia with progeroid features. Although both DM1 and DM2 are characterized by skeletal muscle dysfunction and also share other clinical features, the diseases differ in the muscle groups that are affected. In DM1, distal muscles are mainly affected, whereas in DM2 problems are mostly found in proximal muscles. In addition, manifestation in DM1 is generally more severe, with possible congenital or childhood-onset of disease and prominent CNS involvement. DM1 and DM2 are caused by expansion of (CTG•CAG)n and (CCTG•CAGG)n repeats in the 3' non-coding region of DMPK and in intron 1 of CNBP, respectively, and in overlapping antisense genes. This critical review will focus on the pleiotropic problems that occur during development, growth, regeneration, and aging of skeletal muscle in patients who inherited these expansions. The current best-accepted idea is that most muscle symptoms can be explained by pathomechanistic effects of repeat expansion on RNA-mediated pathways. However, aberrations in DNA replication and transcription of the DM loci or in protein translation and proteome homeostasis could also affect the control of proliferation and differentiation of muscle progenitor cells or the maintenance and physiological integrity of muscle fibers during a patient's lifetime. Here, we will discuss these molecular and cellular processes and summarize current knowledge about the role of embryonic and adult muscle-resident stem cells in growth, homeostasis, regeneration, and premature aging of healthy and diseased muscle tissue. Of particular interest is that also progenitor cells from extramuscular sources, such as pericytes and mesoangioblasts, can participate in myogenic differentiation. We will examine the potential of all these types of cells in the application of regenerative medicine for muscular dystrophies and evaluate new possibilities for their use in future therapy of DM.
Keywords: RNA toxicity; mesoangioblast; muscle stem cell; myoblast; myogenesis; myotonic dystrophy; pericyte; proteotoxicity.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous