

Folliculin Regulates Osteoclastogenesis Through Metabolic Regulation
- PMID: 29893999
- PMCID: PMC6220829
- DOI: 10.1002/jbmr.3477
Folliculin Regulates Osteoclastogenesis Through Metabolic Regulation
Abstract
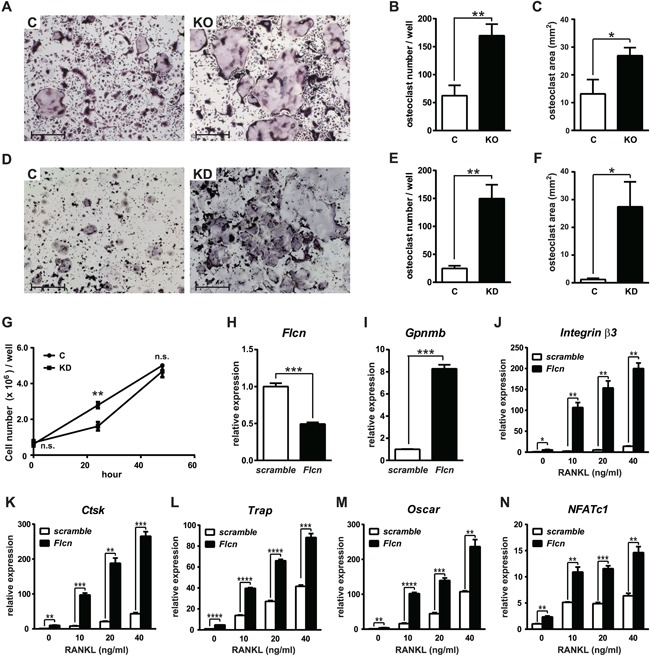
Osteoclast differentiation is a dynamic differentiation process, which is accompanied by dramatic changes in metabolic status as well as in gene expression. Recent findings have revealed an essential connection between metabolic reprogramming and dynamic gene expression changes during osteoclast differentiation. However, the upstream regulatory mechanisms that drive these metabolic changes in osteoclastogenesis remain to be elucidated. Here, we demonstrate that induced deletion of a tumor suppressor gene, Folliculin (Flcn), in mouse osteoclast precursors causes severe osteoporosis in 3 weeks through excess osteoclastogenesis. Flcn-deficient osteoclast precursors reveal cell autonomous accelerated osteoclastogenesis with increased sensitivity to receptor activator of NF-κB ligand (RANKL). We demonstrate that Flcn regulates oxidative phosphorylation and purine metabolism through suppression of nuclear localization of the transcription factor Tfe3, thereby inhibiting expression of its target gene Pgc1. Metabolome studies revealed that Flcn-deficient osteoclast precursors exhibit significant augmentation of oxidative phosphorylation and nucleotide production, resulting in an enhanced purinergic signaling loop that is composed of controlled ATP release and autocrine/paracrine purinergic receptor stimulation. Inhibition of this purinergic signaling loop efficiently blocks accelerated osteoclastogenesis in Flcn-deficient osteoclast precursors. Here, we demonstrate an essential and novel role of the Flcn-Tfe3-Pgc1 axis in osteoclastogenesis through the metabolic reprogramming of oxidative phosphorylation and purine metabolism. © 2018 The Authors Journal of Bone and Mineral Research published by Wiley Periodicals, Inc. on behalf of American Society for Bone and Mineral Research (ASBMR).
Keywords: FOLLICULIN (FLCN); METABOLISM; OSTEOCLAST; OSTEOPOROSIS; TRANSCRIPTION FACTORS.
© 2018 The Authors Journal of Bone and Mineral Research published by Wiley Periodicals, Inc. on behalf of American Society for Bone and Mineral Research (ASBMR).
Figures






References
-
- Ishii KA, Fumoto T, Iwai K, et al. Coordination of PGC‐1beta and iron uptake in mitochondrial biogenesis and osteoclast activation. Nat Med. 2009;15(3):259–66. - PubMed
-
- Nishikawa K, Iwamoto Y, Kobayashi Y, et al. DNA methyltransferase 3a regulates osteoclast differentiation by coupling to an S‐adenosylmethionine‐producing metabolic pathway. Nat Med. 2015;21(3):281–7. - PubMed
-
- Lemma S, Sboarina M, Porporato PE, et al. Energy metabolism in osteoclast formation and activity. Int J Biochem Cell Biol. 2016;79:168–80. - PubMed
-
- Nickerson ML, Warren MB, Toro JR, et al. Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt‐Hogg‐Dube syndrome. Cancer Cell. 2002;2(2):157–64. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
