

Glucocorticoid-dependent REDD1 expression reduces muscle metabolism to enable adaptation under energetic stress
- PMID: 29895328
- PMCID: PMC5998563
- DOI: 10.1186/s12915-018-0525-4
Glucocorticoid-dependent REDD1 expression reduces muscle metabolism to enable adaptation under energetic stress
Abstract
Background: Skeletal muscle atrophy is a common feature of numerous chronic pathologies and is correlated with patient mortality. The REDD1 protein is currently recognized as a negative regulator of muscle mass through inhibition of the Akt/mTORC1 signaling pathway. REDD1 expression is notably induced following glucocorticoid secretion, which is a component of energy stress responses.
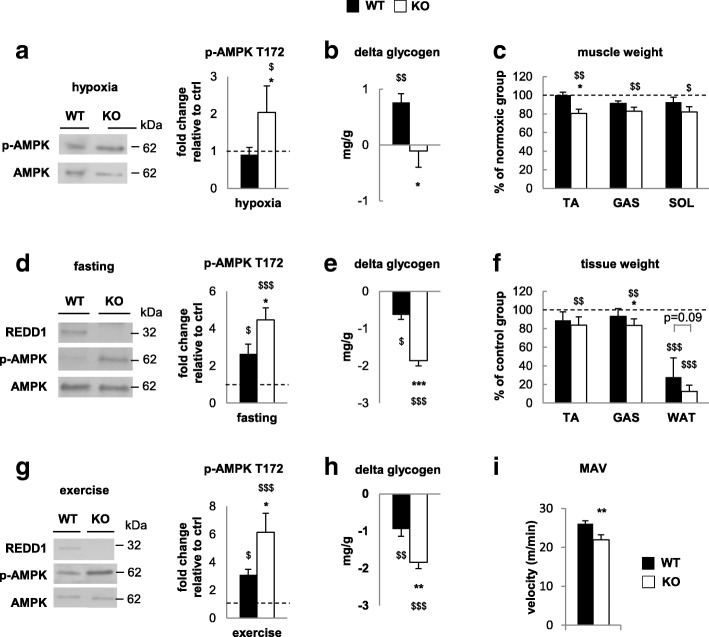
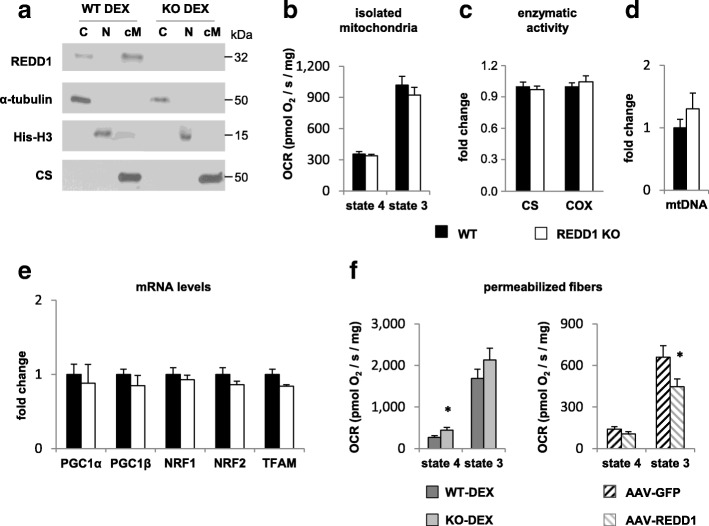
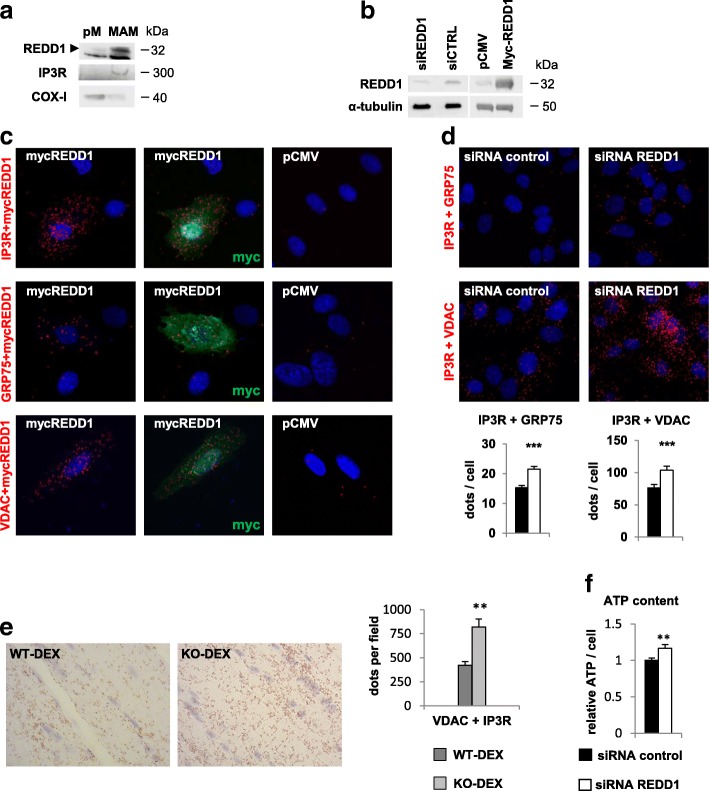
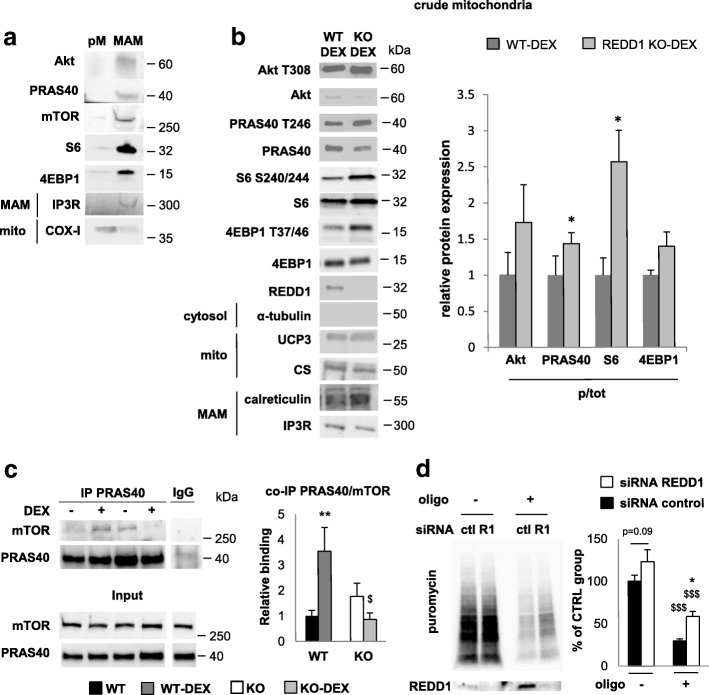
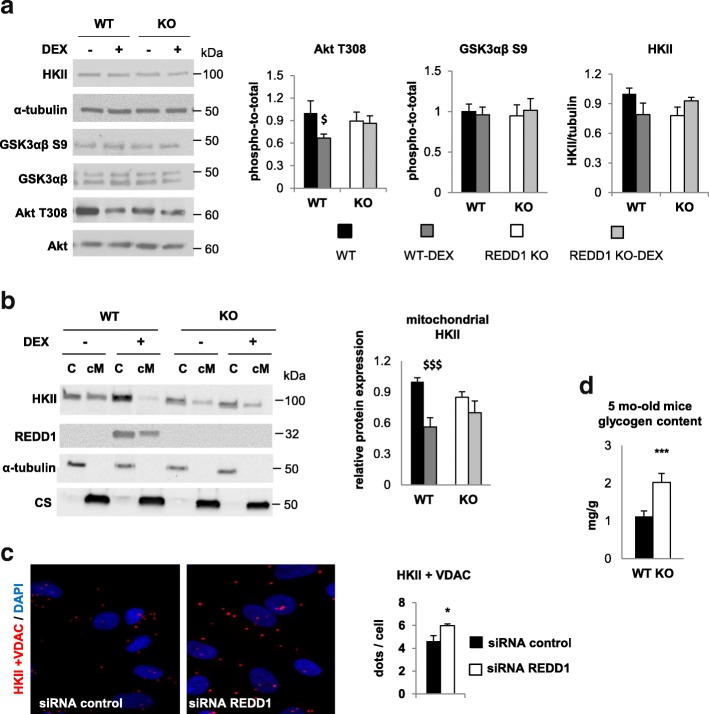
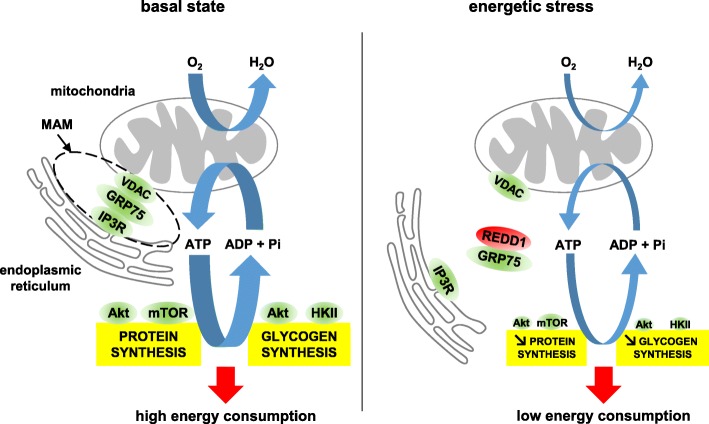
Results: Unexpectedly, we show here that REDD1 instead limits muscle loss during energetic stresses such as hypoxia and fasting by reducing glycogen depletion and AMPK activation. Indeed, we demonstrate that REDD1 is required to decrease O2 and ATP consumption in skeletal muscle via reduction of the extent of mitochondrial-associated endoplasmic reticulum membranes (MAMs), a central hub connecting energy production by mitochondria and anabolic processes. In fact, REDD1 inhibits ATP-demanding processes such as glycogen storage and protein synthesis through disruption of the Akt/Hexokinase II and PRAS40/mTORC1 signaling pathways in MAMs. Our results uncover a new REDD1-dependent mechanism coupling mitochondrial respiration and anabolic processes during hypoxia, fasting, and exercise.
Conclusions: Therefore, REDD1 is a crucial negative regulator of energy expenditure that is necessary for muscle adaptation during energetic stresses. This present study could shed new light on the role of REDD1 in several pathologies associated with energetic metabolism alteration, such as cancer, diabetes, and Parkinson's disease.
Keywords: Energy expenditure; Exercise; Fasting; Hypoxia; MAMs; Metabolism; Mitochondria; Skeletal muscle; mTOR.
Conflict of interest statement
Ethics approval and consent to participate
All procedures on mice comply with European directives (86/609/CEE) and were approved by the national ethics committee (referral file APAFIS#9706-2017042516166776 v3).
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures

References
-
- Shoshani T, Faerman A, Mett I, Zelin E, Tenne T, Gorodin S, Moshel Y, Elbaz S, Budanov A, Chajut A, et al. Identification of a novel hypoxia-inducible factor 1-responsive gene, RTP801, involved in apoptosis. Mol Cell Biol. 2002;22(7):2283–2293. doi: 10.1128/MCB.22.7.2283-2293.2002. - DOI - PMC - PubMed
-
- Ellisen LW, Ramsayer KD, Johannessen CM, Yang A, Beppu H, Minda K, Oliner JD, McKeon F, Haber DA. REDD1, a developmentally regulated transcriptional target of p63 and p53, links p63 to regulation of reactive oxygen species. Mol Cell. 2002;10(5):995–1005. doi: 10.1016/S1097-2765(02)00706-2. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
